
液液萃取-高分辨气相色谱-高分辨双聚焦磁质谱法测定尿中羟基多环芳烃代谢物
2020-04-22 00:29陆一夫胡小键杨艳伟
色谱 2020年6期
付 慧,陆一夫,胡小键,张 续,杨艳伟,朱 英
(中国疾病预防控制中心环境与健康相关产品安全所, 北京 100021)
多环芳烃(PAHs)来源于各种矿物燃料(如煤、石油、天然气等)及有机物的热解或不完全燃烧,在空气、水、土壤等各种环境介质中广泛存在[1]。尤其是现代工业的兴起,飞机、汽车等各种机动车辆日益增加及其他人为排放,导致PAHs的污染日益严重。人们在日常生活中,通过呼吸、饮食、饮水,甚至皮肤接触均会不同程度地暴露于PAHs[2-5]。研究表明,PAHs暴露可损害动物中枢神经,破坏淋巴细胞微核率,肝脏功能和DNA修复能力,是具有“三致”毒性的化学物质。此外,PAHs还具有内分泌干扰效应,可影响生殖内分泌系统功能,并导致遗传损害[1]。PAHs被认为是数量最多、分布最广、与人类关系最密切、对人体健康威胁最大的环境致癌物之一,多环芳烃的人群暴露问题已逐渐成为日益重视的课题。
进入体内的PAHs在肝脏中分两步转化为结合态单羟基多环芳烃(OH-PAHs),随后经尿液或粪便排出体外[6]。由于PAHs在体内代谢期较短,且尿液最易获得和保存,人体尿液中OH-PAHs的分析被用作评估人类接触PAHs最常用的方法。联合多个PAHs代谢物作为生物标志物,能更全面评估人体PAHs暴露水平。因此,建立快速灵敏、准确稳定的同时测定尿液中多种OH-PAHs的方法对于PAHs的暴露评价具有重要意义。
目前,测定尿中OH-PAHs常用的方法有气相色谱-串联质谱法(GC-MS/MS)[7-10]、高效液相色谱-荧光检测法(HPLC-FLD)[11-13]和液相色谱-串联质谱法(LC-MS/MS)[14-17]。国内研究者测定尿中OH-PAHs多采用LC-MS/MS,国外研究者多采用GC-MS/MS。本实验室前期也利用LC-MS/MS建立了尿中12种OH-PAHs的测定方法,但基质效应会影响测定结果的准确性。采用气相色谱作为分离系统,虽较液相色谱多了衍生化步骤,但其具有较低的基质效应和稳定的色谱保留时间,适合复杂基质的超痕量分析。串联质谱虽具有较好的选择性和灵敏度,但其分辨率不够高,对相对分子质量较高的离子有质量歧视效应[18];高分辨双聚焦磁质谱具有高质量分辨率和高质量精度(质量偏差通常≤10-6),能为复杂样品基质中痕量物质的精准检测提供有力的技术保障[19,20]。

1 实验部分
1.1 仪器、试剂与材料
Autospec Premier高分辨双聚焦磁质谱仪(EI源,美国Waters Micromass公司), 7890A气相色谱仪(美国Agilent公司), T-214电子天平(精度0.1 mg,美国Denver公司), LW-20水浴摇床(美国LabTech公司), EFCG11848氮吹仪(美国Organomation公司), GZX-9240MBE数显鼓风干燥箱(上海博迅实业有限公司医疗设备厂), 600C离心机(北京白洋医疗器械有限公司)。

正戊烷(GC级,德国Merck公司),甲苯(HPLC级,美国Tedia公司),十二烷(纯度99%,比利时Acros公司),抗坏血酸(德国CNW公司),N-甲基-N-(三甲基硅烷)三氟乙酰胺(MSTFA,纯度≥98.5%,美国Sigma-Aldrich公司),β-葡萄糖苷酸酶/硫酸芳酯酶混合酶(β-葡萄糖苷酸酶30 U/mL、硫酸芳酯酶60 U/mL)、超纯水(德国Merck公司),标准参考物质SRM3672(美国NIST标准物质)。
1.2 标准溶液配制
准确称取一定量的1-OHN标准品,用甲醇溶解并定容至10 mL,配制成质量浓度约为2 mg/mL的单标储备液。各取一定体积的1-OHN单标储备液和7种化合物的混合标准溶液,用甲苯溶解并定容至10 mL,配制成1-OHN质量浓度为5.0 mg/L、其他各组分质量浓度为1.0 mg/L的混合标准中间液。用甲苯逐级稀释配制得混合标准使用液。
称取一定量的内标标准品,用甲醇溶解并定容至10 mL,配制成质量浓度约为2 mg/mL的单标储备液。量取一定体积的8种单标储备液,混合,用甲苯配制成各组分质量浓度均为5 mg/L的混合标准中间液;用甲苯逐级稀释得到质量浓度为200 μg/L的混合内标使用液。
回收标准溶液(RSS):用甲苯稀释13C-PCB105壬烷溶液至质量浓度为100 ng/mL,用棕色瓶分装,于4 ℃保存备用。
1.3 实验方法
1.3.1样品前处理
将冰冻尿样置于室温,移取2 mL尿样,置于12 mL玻璃离心管中,分别加入1 mL醋酸钠溶液(1 mol/L, pH=5.5)、20 μL抗坏血酸溶液(250 mg/mL)、50 μLβ-葡萄糖苷酸酶/硫酸芳酯酶和20 μL 200 μg/L的混合内标使用液,混匀,于37 ℃水浴条件下避光水解约17 h,得水解液。
水解液中加入2 mL去离子水和5 mL甲苯-戊烷(1∶4, v/v)萃取剂,振摇30次,以3 600 r/min离心8 min,收集上层有机相;加入5 mL萃取剂,重复萃取,合并两次萃取液。萃取液中加入1 mL 1 mol/L硝酸银溶液,振摇30次,以3 600 r/min离心8 min,将上层有机相转移至氮吹试管中,加入20 μL十二烷,于40 ℃、5 Pa氮吹20 min,使戊烷挥发;升温至70 ℃,使甲苯挥发至近干。向氮吹试管中加入20 μL甲苯复溶,转移至已加入20 μL RSS和100 μL衍生化试剂MSTFA的棕色进样小瓶中,混匀,于60 ℃烘箱中衍生50 min,待测。
1.3.2色谱条件
色谱柱:DB-5MS气相色谱柱(30 m×0.25 mm×0.25 μm);进样方式:不分流进样(使用填充玻璃棉的超惰性内衬管,最大限度减少柱污染);进样口温度:270 ℃;传输管温度:270 ℃;升温程序:初始温度95 ℃,保持2 min,以15 ℃/min升温至160 ℃,再以5 ℃/min升温至205 ℃,保持6 min,然后以10 ℃/min升温至225 ℃,再以29 ℃/min升温至310 ℃,保持6 min;载气:高纯氦气,恒定流量1.0 mL/min;进样量:1.0 μL。
1.3.3质谱条件
离子源:EI源;离子源温度:260 ℃;单离子接收检测(voltage SIR);电子能量:35 eV;捕集电流:650 μA;光电倍增电压:350 V;分辨率:10 000。8种OH-PAHs及其同位素内标的质谱参数见表1。
1.4 质量控制
羟基多环芳烃属于光敏性物质,实验区域应使用防紫外线的荧光灯,以减少其阳光暴露。每批样品检测应附带质控样品、空白样品,以保证方法的准确性并及时监控空白污染。随机抽取未知样品总数的10%进行重复测定,以保证方法的精密度,重复测定样品的相对偏差不应超过30%。目标物相对于内标的相对保留时间比不能偏离校准标准的0.15%以上,每批样品RSS的相对标准偏差不大于10%。
2 结果与讨论
2.1 色谱条件优化
实验通过梯度升温程序,使不同组分得到了较好分离,且峰形尖锐、对称。在自动取样器取样前、后增设多次洗针程序,以消除连续取样过程引起的目标物残留。8种OH-PAHs和8种相应同位素内标的色谱图见图1。
2.2 酶及其用量的选择
根据前期经验[14]及文献报道[21,22],比较了单一β-葡萄糖苷酸酶、β-葡萄糖苷酸酶/硫酸酯酶混合酶、固体β-葡萄糖苷酸酶/硫酸芳酯酶混合酶、液体β-葡萄糖苷酸酶/硫酸芳酯酶混合酶共4类酶对SRM3672质控样品的酶解效果。结果表明,使用固体和液体β-葡萄糖苷酸酶/硫酸芳酯酶混合酶的测定结果较理想。其中,固体酶在使用时需要超声溶解,容易出现溶解不充分的现象,而液体酶使用方便。故本研究最终选用液体β-葡萄糖苷酸酶/硫酸芳酯酶混合酶进行酶解。
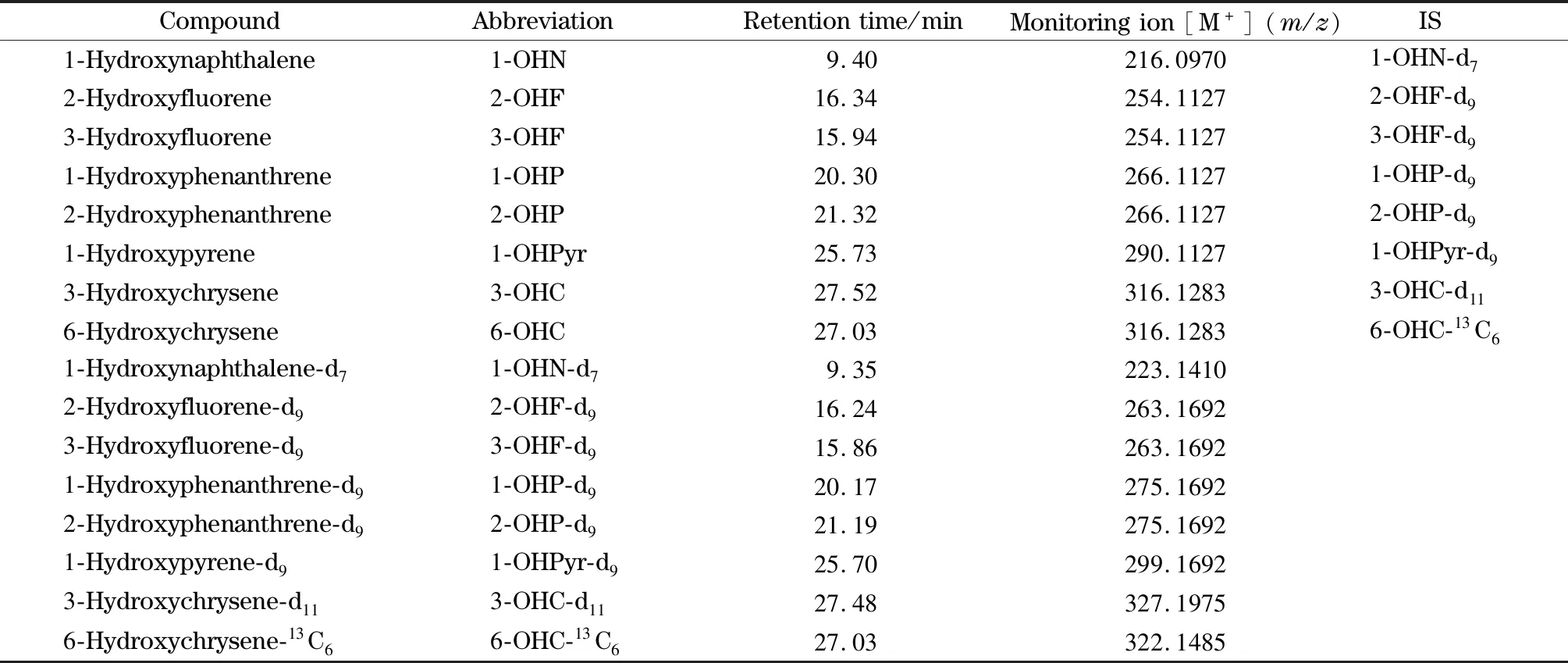
表 1 8种OH-PAHs及其同位素内标的保留时间和质谱参数

图 1 8种OH-PAHs及其同位素内标的色谱图Fig. 1 Chromatograms of the eight OH-PAHs and their isotope internal standardsMSTFA: N-methyl-N-(trimethylsilyl)trifluoroacetamide.
在2 mL实际尿样中加入不同体积的液体混合酶,通过考察目标物的响应值,确定酶的最佳使用量。最终确定2 mL尿样中需加入50 μL液体混合酶。
2.3 衍生化试剂用量的选择
衍生化是高分辨气相色谱-高分辨双聚焦磁质谱分析OH-PAHs的一个重要环节。实验在高浓度标准溶液中加入不同体积的衍生化试剂MSTFA,通过考察目标物的响应值,确定MSTFA的合理使用量(见图2)。在20 μL混合标准溶液(1-OHN 4 mg/L,其他组分0.8 mg/L)中,分别加入10、20、40、80、100和120 μL的MSTFA,随着MSTFA用量的增加,目标物的响应值逐渐增大,当用量达到100 μL时,响应趋于稳定,且色谱图中出现MSTFA的色谱峰(保留时间9.55 min,m/z223.141 0),说明MSTFA过量,反应完全。最终确定MSTFA的加入量为100 μL。

图 2 不同衍生化试剂用量对目标化合物响应值的影响Fig. 2 Effect of different amount of derivatization reagents on the response values of the target compounds

表 2 8种OH-PAHs的线性范围、回归方程、相关系数、检出限和定量限
y: peak area ratio of quantitative ion of the analyte to IS;x: mass concentration ratio of the analyte to IS.
2.4 方法学考察
2.4.1线性范围和检出限
以甲苯为溶剂,配制质量浓度分别为0.83、4.17、8.33、41.7、83.3、417和833 μg/L(1-OHN的质量浓度为上述质量浓度的5倍),内标质量浓度为33.3 μg/L的系列单标标准溶液(折算到人尿中的质量浓度分别为0.008、0.042、0.083、0.417、0.833、4.17和8.33 μg/L, 1-OHN的浓度为其的5倍)。各取20 μL上述单标标准溶液,转移至已加入20 μL RSS和100 μL MSTFA的棕色进样小瓶中,混匀,于60 ℃烘箱中衍生50 min后测定。对目标物与相应内标离子峰的峰面积之比(y)和目标物与相应内标质量浓度之比(x)进行线性回归分析,绘制校准曲线,得到8种目标化合物的线性方程和相关系数(r)。结果显示,8种OH-PAHs的线性关系良好,r均>0.99(见表2)。参照美国环保署(US EPA)检出限测定程序文件第2版[23],分别采用两种方法(第一种考虑方法空白,计算6个月内运行的方法空白平均值加3倍标准偏差;第二种考虑仪器灵敏度,计算3倍信噪比)考察检出限,并取其高值作为方法的检出限,再以3.3倍检出限作为定量限。
考虑到样品经前处理后浓缩100倍,本方法对人尿中8种OH-PAHs的方法检出限为0.006~0.042 μg/L,定量限为0.020~0.140 μg/L,结果见表2。
2.4.2加标回收率和相对标准偏差
以实际尿样进行高、中、低3个水平的8种OH-PAHs的加标回收试验,加标水平分别为0.125、1.25和6.25 μg/L,每个水平进行6次平行试验,计算加标回收率。8种OH-PAHs的平均回收率为81.4%~127.0%。在同一天及不同天对实际尿液样品进行分析测定(n=6),以确定方法的日内精密度和日间精密度。结果表明,8种OH-PAHs的日内和日间精密度分别为2.7%~6.9%和5.1%~10.9%(见表3)。
2.4.3准确性验证
通过美国NIST标准参考物质SRM3672进一步评价该方法的准确性。用该方法对SRM3672进行检测,并与其认证的6种物质的浓度进行比较。OH-PAHs的认证浓度来自SRM3672的分析证书,并将分析证书中的含量(μg/kg)通过尿密度(1.019 g/mL)转换成质量浓度(μg/L)。如表4所示,该方法对OH-PAHs的检测结果与经过认证的浓度一致,且包含不同数量级,进一步证明了该方法的准确性。

表 3 8种OH-PAHs在尿样中的加标回收率和日内、日间精密度(n=6)

表 4 SRM3672平均测定含量与认证含量(n=6)
NIST: National Institute of Standards and Technology.
2.5 实际样品测定
对某地区330份实际人群尿样进行检测,以了解该地区人群PAHs暴露水平。受试者被告知研究目的并签署知情同意书。结果显示,1-OHN、2-OHF、3-OHF、1-OHP、2-OHP和1-OHPyr的检出率为100%, 3-OHC和6-OHC未检出。1-OHN的检出范围为0.237~12.4 μg/L,2-OHF为0.021~4.0 μg/L,3-OHF为0.010~1.4 μg/L,1-OHP为0.025~4.6 μg/L,2-OHP为0.020~0.85 μg/L和1-OHPyr为0.010~1.4 μg/L。
3 结论
本文建立了液液萃取-高分辨气相色谱-高分辨双聚焦磁质谱测定人体尿样中8种OH-PAHs的分析方法。本方法前处理成本低,选择性好,灵敏度高,结果可靠,对于相对分子质量较低的PAHs代谢物的分析有明显优势,在人群生物监测工作中有很好的应用价值。
猜你喜欢
家庭科学·新健康(2022年2期)2022-03-07
中华养生保健(2020年9期)2021-01-18
石油化工技术与经济(2020年4期)2020-09-15
无机化学学报(2019年2期)2019-02-27
天然产物研究与开发(2018年8期)2018-09-10
天然产物研究与开发(2018年4期)2018-05-07
分析化学(2017年12期)2017-12-25
环球时报(2016-05-18)2016-05-18
中国环境科学(2015年7期)2015-01-28
郑州大学学报(工学版)(2014年6期)2014-03-01
