
核苷酸结合寡聚化结构域样受体蛋白3炎性小体抑制剂
2020-04-08 01:24:20兀娜沈敏
中华临床免疫和变态反应杂志 2020年1期
兀娜,沈敏
炎性小体是一类模式识别受体(pattern recognition receptors,PRRs),可识别细胞内病原相关分子模式(pathogen associated molecular patterns,PAMPs)或损伤相关分子模式(damage associated molecular patterns,DAMPs),是固有免疫的重要组成成分。NLRP3炎性小体是目前研究最广泛、最明确的一种炎性小体,由核苷酸结合寡聚化结构域(nucleotide-binding oligomerization domain,NOD)样受体蛋白3(Nod-like receptor protein3, NLRP3)、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing CARD,ASC)和半胱氨酸天冬氨酸蛋白酶 1 (cysteine-requiring aspartate protease-1,caspase-1)组成(图1)。NLRP3与PAMPs或DAMPs结合后招募下游分子ASC,将无活性的促caspase-1前体活化为有生物学功能的caspase-1,caspase-1能够剪切焦亡蛋白 GasderminD触发细胞焦亡,并且促进了白介素 (interleukin,IL)-1β和IL-18等炎性细胞因子的成熟和分泌。IL-1β和IL-18作为强炎性因子能够有效促进免疫反应,过表达时会造成急慢性炎性损伤,NLRP3炎性小体及其下游分子在一系列炎症性疾病的发生发展过程中扮演了重要的角色,如2型糖尿病、阿尔兹海默病、痛风、NLRP3相关自身炎症性疾病(NLRP3-associated autoinflammatory disease, NLRP3-AID)(以往称为冷炎素相关周期性综合征,cryopyrin-associated periodic syndrome,CAPS)、帕金森病等[1-5]。NLRP3炎性小体的激活有以下4种可能的机制(图1):(1)胞外三磷酸腺苷(adenosine triphosphate, ATP)刺激细胞膜上的P2X7型嘌呤受体,引起钾离子选择性通道开放,胞内钾离子浓度降低是NLRP3炎性小体激活的一个重要因素[6];(2)晶体物质被细胞吞噬后破坏溶酶体膜稳定性,溶酶体破裂释放出组织蛋白酶B(cathepsinB,CB),致使NLRP3炎性小体激活[7];(3)有氧代谢的副产物活性氧(reactive oxygen species,ROS)从线粒体释放,或线粒体损伤释放的线粒体DNA均被证实能够促进NLRP3炎性小体的激活[8-9];(4)NLRP3通过其保守的碱性区域与带负电荷的磷脂酰肌醇-4-磷酸(phosphatidylinositol-4-phosphate,PtdIns4P)结合而被募集到分散高尔基体反面网络结构(dispersed trans-Golgi net-work,dTGN),后者能够提供结构支点促使NLRP3招募ASC以完成小体的组配[10]。通过抑制NLRP3炎性小体的激活可以治疗NLRP3炎性小体相关疾病,因此NLRP3炎性小体抑制剂成为近年来的研究热点。本文按照不同作用靶点,对NLRP3炎性小体抑制剂的最新研究进展进行综述。
1 以NLRP3为作用靶点
1.1 阻断NLRP3中心区域ATP酶活化
NLRP3主要由3部分构成:碳端亮氨酸富集结构域(LRRs),可识别并结合PAMPs和DAMPs;中心区域存在ATP酶活化有关的核苷酸寡寡聚结合域,称为NBD或NACHT;氮端为效应结构域即pyrin结构域(PYD),负责招募下游信号分子。其中NACHT区域ATP酶的活化是NLRP3寡聚化以及ASC招募的重要步骤[11],阻断NLRP中心区域ATP酶活化可抑制NLRP3炎性小体活化。
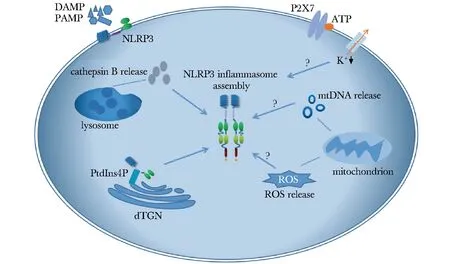
图1NLRP3炎性小体激活机制
Fig1Activation mechanism of NLRP3 inflammasome
NLRP3:核苷酸结合寡聚化结构域样受体蛋白3;DAMPs:损伤相关分子模式;PAMPs:病原相关分子模式;lysosome:溶酶体;Cathepsin B release:组织蛋白酶B释放;dTGN:分散高尔基体反面网络结构;PtdIns4P:磷脂酰肌醇-4-磷酸;mitochondrion:线粒体;ROS release:活性氧释放;mtDNA release:线粒体DNA释放;P2X7:胞外核苷酸P2X7型受体;ATP:三磷酸腺苷;K+:钾离子;NLRP3 inflammasome assembly:NLRP3炎性小体组装
1.1.1 OLT1177
活性β-磺酰腈化合物OLT1177是氯化剂和甲硫氨酸合成反应产物的活性成分,能负性调控NACHT结构域ATP酶结合位点,抑制NLRP3炎性小体活化。免疫沉淀反应以及荧光能量共振转移分析技术表明OLT1177还能够阻断NLRP3与ASC和caspase-1的相互作用。OLT1177对NLRC4和黑色素瘤缺乏因子2(absent in melanoma2,AIM2)炎性小体无作用,也不影响钾离子外流或IL-1β前体的合成,因此该药物是一种特异性NLRP3炎性小体抑制剂。Marchetti等[12]研究发现,体外实验中OLT1177可以抑制小鼠巨噬细胞产生IL-1β、减少人外周血中性粒细胞和单核细胞产生IL-1β、抑制caspase-1活化;小鼠体内实验发现OLT1177能够减轻脂多糖(LPS)诱发的全身炎症。对痛风性关节炎及酵母聚糖诱发的急性反应性关节炎小鼠模型应用OLT1177可改善关节腔积液中炎细胞浸润情况。该药用于治疗痛风性关节炎的I期临床试验已取得成功,未发现OLT1177相关的显著不良事件,现已进入临床II期试验[13]。
1.1.2 CY-09
Jiang等[14]发现小分子化合物CY-09(分子式:C19H12F3NO3S2)可以与ATP竞争结合NACHT结构域上的与ATP酶活化有关的的Walker A基序,减少ATP酶的活化进而阻断NLRP3炎性小体激活。CY-09不影响NLRP3蛋白的泛素化、钾离子外流等,并且仅作用于NLRP3炎性小体,对NLRC4、NLRP1等炎性小体没有作用,是NLRP3炎性小体特异性抑制剂。CY-09治疗NLRP3-AID和2型糖尿病小鼠模型均有显著效果,且具有较好的安全性、稳定性及口服生物活性[14],但目前尚未进行人体试验。
1.2 抑制NLRP3去泛素化
NLRP3的转录后修饰是调控其活化的关键步骤之一[15]。去泛素化酶(deubiquitinating enzymes,DUB)BRCC3可使NLRP3的LRR结构域去泛素化,该环节在NLRP3炎性小体活化过程中起到了关键作用[16]。
1.2.1 G5
G5(分子式:C19H14N2O7S)是一种小分子DUB抑制剂,也称异肽酶抑制剂。2006年有研究首次发现1 μmol G5即可抑制LPS-ATP诱导的小鼠巨噬细胞分泌IL-1β,但具体机制不详[17]。Py等[18]进一步证实G5通过负性调控NLRP3去泛素化而抑制NLRP3炎性小体激活,阻断caspase-1活化,并不作用于NLRC4及AIM2 炎性小体,因此是另一种特异性NLRP3炎性小体抑制剂。然而,也有学者认为DUB抑制剂是通过破坏NLRP3炎性小体中ASC的寡聚化和ASC斑点形成而抑制NLRP3炎性小体活化[19]。
1.2.2 多巴胺
多巴胺(dopamine,DA)是中枢神经系统内一种重要的神经递质,近年来有学者研究揭示了DA也参与了免疫反应过程。多数DA受体表达于免疫细胞表面,包括巨噬细胞和树突样细胞,DA与细胞表面的受体结合发挥效应。现已发现的DA受体有5个亚型:DRD1,DRD2,DRD3,DRD4及DRD5。2014年Yan等[20]发现DA是一种内源性NLRP3小体抑制剂,对单钠尿酸盐(mono-sodium urate,MSU)、明矾、尼日利亚菌素以及ATP诱导的NLRP3活化均表现出抑制作用;由DA激活的DRD1信号通路不仅可以缓解小鼠体内神经毒素MPTP诱导的神经炎症,还能改善 LPS诱导的外周炎症以及MSU诱导的腹腔感染。在DA发挥抑制NLRP3小体作用过程中,DRD1起到了主要作用,DRD5作用次之,而DRD2、DRD3和DRD4则未发挥作用。在DRD1基因敲除(Drd1-/-)小鼠的巨噬细胞内,DA则未能发挥抑制NLRP3炎性小体的作用,因此进一步证实了DRD1的作用。
DA通过DRD1信号通路改变了腺苷酸环化酶(cAMP)构型,使其活性增强,cAMP的生成增多,小鼠巨噬细胞内cAMP活化E3泛素连接酶MARCH7,催化NLRP3的泛素化使其经细胞自噬过程被降解。这与cAMP激活剂Forskolin能够负性调控NLRP3炎性小体的活化所示的机制类似[21]。上述研究结果提示DA可能是治疗NLRP3炎性小体相关性疾病的一种有效药物。
1.3 抑制NLRP3与NLRP3的连接
抗过敏药物曲尼司特(Tranilast,TR)能够抑制IgE介导肥大细胞释放组胺的过程,临床应用于治疗和预防过敏性哮喘及过敏性皮炎已有近40年的历史,安全性良好。Huang等[22]发现TR能够直接与NLRP3结合进而抑制NLRP3炎性小体的活化,与OLT1177、CY-09不同的是,TR对NLRP3炎性小体的抑制作用与NACHT结构域的ATP酶活性无关,而是直接影响NLRP3与NLRP3的相互连接,进而阻断了NLRP3炎性小体的组装。痛风性关节炎及NLRP3-AID小鼠模型应用TR后,体内IL-1β、IL-18浓度低于空白对照组,关节腔积液量减少,死亡率也明显减低;痛风性关节炎患者关节滑液细胞加入TR共培养后,caspase-1活化和IL-1β生成减少,且与TR的剂量存在相关性。后文即将提及的MCC950是公认的NLRP3炎性小体强效特异性抑制剂,TR与MCC950的对照试验表明TR在体外的药物效能约是MCC950的1/400,但在体内其药物效能约为MCC950的1/10~1/5,且TR不影响NLRC4或AIM2炎性小体,对钾离子外流及线粒体损伤无作用,特异性较强。TR的相关研究为探索老药的新作用提供了重要思路。
2 以ASC为作用靶点
NLRP3依赖性的ASC寡聚化被认为是NLRP3炎性小体激活的关键环节,ASC寡聚化后形成caspase-1活化的平台即ASC斑点[23-24]。因此,抑制ASC寡聚化可抑制NLRP3炎性小体活化。
2.1 MCC950
2001年Perregaux等[25]首次发现二芳基磺酰脲复合物有抑制IL-1β分泌的功能,其中,MCC950被研究得最为深入,是一种公认的强效特异性小分子NLRP3炎性小体抑制剂。MCC950通过抑制ASC的寡聚化和ASC斑点形成特异性地抑制NLRP3炎性小体的活化,低浓度的MCC950即能抑制尼日利亚菌素诱导的小鼠体内骨髓来源巨噬细胞和外周血单核细胞释放IL-1β,该药不影响钾离子外流,也不作用于NLRC4、AIM2炎性小体。MCC950在NLRP3-AID小鼠模型体内及从NLRP3-AID患者体内提取的外周血单核细胞中均能有效抑制NLRP3炎性小体,体内/外药代动力学实验表明其口服有效生物利用率高达68%[26]。
MCC950在除NLRP3-AID之外的其他疾病也可能发挥作用。帕金森病是一种常见的神经退行性病变,α-突触核蛋白是与该病相关的基因突变编码蛋白,可以激活NLRP3炎性小体[5]。MCC950能够通过血脑屏障,且口服有效,该药可阻断临床前期帕金森病小鼠模型α-突触核蛋白原纤维诱导的ASC寡聚化,下调NLRP3炎性小体的表达,最终减缓小鼠中枢神经系统黒质纹状体多巴胺能神经元损害[27]。
Hout等[28]应用MCC950治疗心肌梗死猪模型(3~6 mg/kg,共7 d),MCC950组与对照组相比,实验动物血清IL-1β浓度降低,左室射血分数保留度高,通过伊文氏蓝/TTC染色确定风险区域(area at risk,AR)及梗死面积(infarct size,IS)发现前者的AR/IS值低于后者,提示MCC950具有改善心肌梗死预后的潜能。此外,对颈动脉粥样硬化小鼠模型应用MCC950(10 mg/kg,3次/周,共4周)后观察到小鼠颈部血管粥样斑块体积减小,血清IL-1β及血管细胞黏附分子(VCAM-1)浓度减低[29]。
2.2 β-羟丁酸
内源性化合物β-羟丁酸(β-hydroxybutyrate,BHB)可阻断ATP介导的ASC寡聚化及ASC斑点形成,部分抑制巨噬细胞内钾离子的外流,从而负性调控炎性小体的装配。由于BHB在人体内的清除率较高,故常常将BHB和纳米脂质体聚合凝胶(nanolipogel, nLGs)同时使用以提高其在血液中的浓度以及生物活性。对注射MSU的小鼠应用nLGs-BHB后,小鼠巨噬细胞产生的IL-1β较对照组明显减少。对NLRP3-AID小鼠模型以及MSU诱导腹膜炎小鼠模型应用nLGs-BHB后均发现caspase-1活化降低,IL-1β产生减少[30]。
3 caspase-1抑制剂
VX-765是一类口服有效的caspase-1抑制剂,在小鼠体内经肝酶作用转化为具有生物代谢活性的VRT-043198。Wannamaker等[31]发现VX-765可显著降低健康小鼠血浆中LPS诱导释放的IL-1β浓度,在剂量为100 mg/kg时效果最为显著;对类风湿关节炎模型小鼠应用VX-765(100 mg/kg,2次/d)后,关节的炎症评分降低,与泼尼松龙(5 mg/kg)疗效相当。
目前普遍认为神经元细胞内β-淀粉样蛋白(amyloid-β,Aβ)聚集沉积形成斑块是阿尔兹海默病的特征性病理学表现,而NLRP3炎性小体被证实参与到Aβ的聚集过程中[2],对阿尔兹海默病小鼠模型应用VX-765(3次/周,1~3周)后,小鼠颅内Aβ沉积减少,瞬时和长期记忆损伤的症状有所改善。淀粉样前体蛋白(Aβ的前体物质)转染的人类神经元细胞加入VX-765共培养可减少神经细胞轴突的串珠状改变[32]。以上发现提示VX-765具有治疗NLRP3炎性小体相关性疾病的潜在可能。
4 其他药物
4.1 铜离子螯合剂四硫钼酸盐
细胞内活性铜离子参与了人和小鼠巨噬细胞内NLRP3炎性小体活化过程[33]。适量的铜离子螯合剂能够减轻LPS诱导的炎性反应[34]。铜离子螯合剂四硫钼酸盐(tetrathiomolybdate,TTM)作为一类有效的铜离子螯合剂,目前在临床上用于治疗Wilison病及乳腺癌,安全性良好。超氧化物岐化酶(super-oxide dismutase 1,SOD1)是一种含铜生物酶,铜是酶分子的活性中心结构并在催化反应中传递电子,参与细胞内抗氧化过程继而降低胞内ROS浓度,而线粒体来源的ROS被认为是调控 NLRP3 炎性体活化的关键信号之一[9]。应用TTM后进行流式细胞分析发现,经LPS诱导的小鼠巨噬细胞ASC含量降低,成熟caspase-1的表达量减少,小鼠的内毒素中毒症状也有所缓解。TTM不作用于AIM2、NLRC4及NLRP1炎性小体,不抑制NF-κB及MAPK通路,具有较强的选择性。同时,还有研究证实TTM能够抑制由铜-Aβ复合体导致氧化应激而产生的ROS,因而可能具有治疗阿尔茨海默病的作用,进一步药理实验还在进行中[35]。
4.2 ω-3多不饱和脂肪酸
有研究发现ω-3多不饱和脂肪酸 ( ω-3 polyunsaturated fatty acid,ω-3PUFA) 能够抑制包括IL-1β在内的炎性细胞因子的产生,但具体机制尚不明确[36]。另一项动物实验结果表明血浆中ω-3 PUFA 含量增高可以显著降低2型糖尿病的发生风险[37]。在此基础上,Yan等[38]研究证实ω-3 PUFA抑制了NLRP3依赖性caspase-1的激活和IL-1β的分泌。G蛋白偶联受体(G protein-coupled receptor,GPR)120和GPR40与ω-3 PUFA的抗炎作用有关[39],β-arrestin-2 (ARRB2) 是GRP120的下游支架蛋白[40],实验发现ω-3 PUFA可以促进ARRB2和NLRP3蛋白结合,间接激活GPR120和GPR40独立信号通路,下调NLRP3炎性小体的表达。2型糖尿病大鼠模型应用ω-3 PUFA后其胰岛素抵抗明显改善,血糖控制趋于稳定。
5 总结与展望
NLRP3炎性小体在多种炎症性、代谢性疾病中发挥了重要作用,以NLRP3炎性小体为靶点治疗NLRP3炎性小体相关性疾病具有广阔的临床前景(图2)。本文所述NLRP3炎性小体抑制剂大多尚未开展临床实验或正式应用于临床。目前仍有许多亟待解决的问题,如NLRP3炎性小体抑制剂的安全性如何评估,能否寻找到不同刺激剂激活NLRP3炎性小体的共同通路并加以抑制,能否联合应用不同类型的NLRP3炎性小体抑制剂等。深入探究NLRP3活化和调控机理并以此为基础研发特异性NLRP3炎性小体抑制剂,为治疗NLRP3炎性小体相关性疾病带来了新的希望。
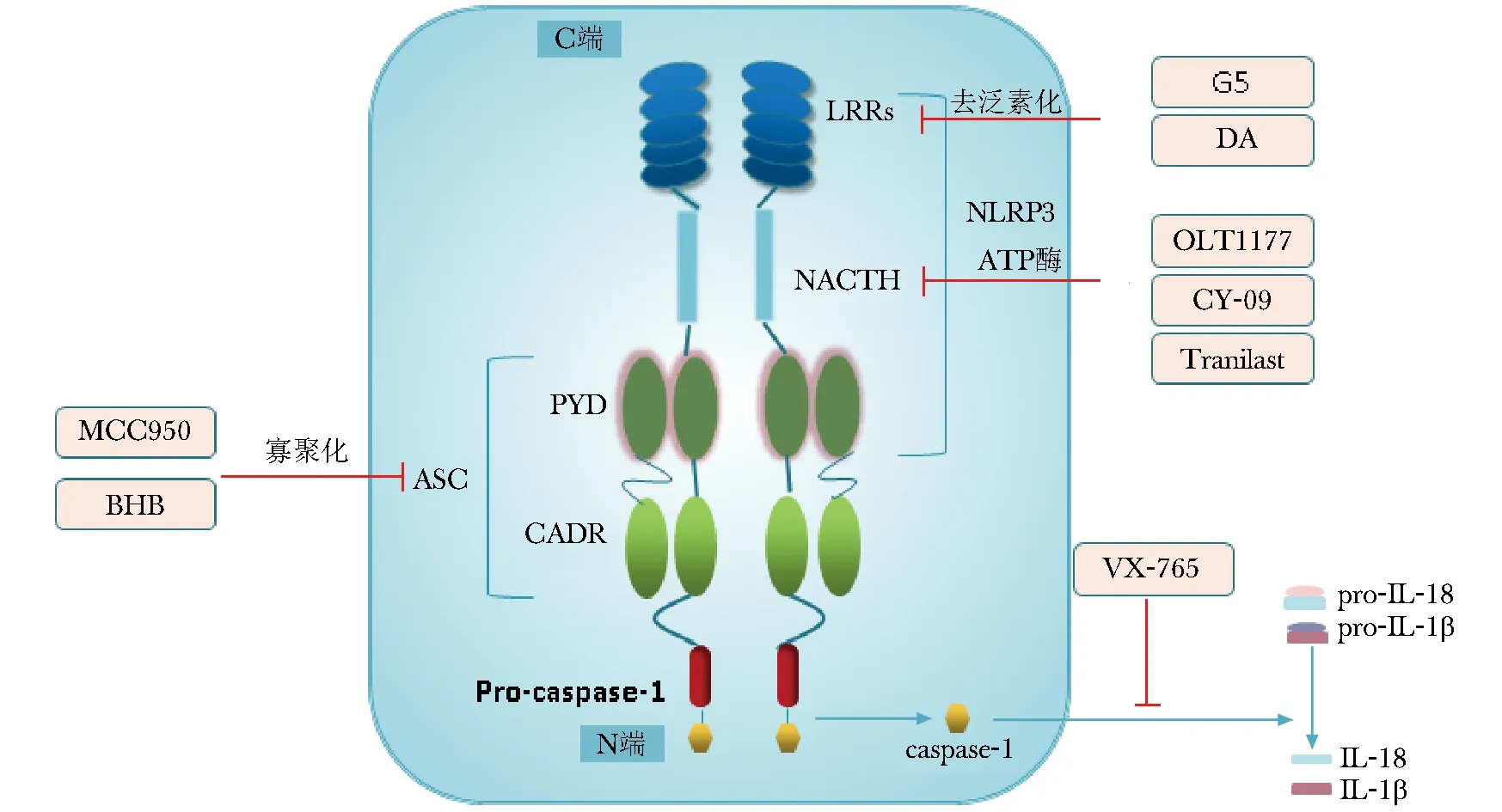
图2NLRP3炎性小体结构示意图及不同抑制剂作用部位
Fig2Structure of NLRP3 inflammasome and targets of different inhibitors
注:NLRP3:核苷酸结合寡聚化结构域样受体蛋白3;ASC:凋亡相关斑点样蛋白;LRRs:亮氨酸富集结构域;NACHT:ATP酶活化有关的核苷酸寡聚结合域;CARD:caspase募集结构域;PYD:pyrin结构域;Pro-caspase-1:半胱氨酸天冬氨酸蛋白酶1前体;caspase-1:半胱氨酸天冬氨酸蛋白酶1;DA:多巴胺;Tranilast:曲尼司特;BHB:β-羟丁酸;pro-IL-18:白介素18前体;pro-IL-1β:白介素1β前体;IL-18:白介素18;IL-1β:白介素1β
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:43
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
西南军医(2015年3期)2015-04-23 07:28:32
国际心血管病杂志(2015年5期)2015-02-27 12:11:34
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
现代检验医学杂志(2014年6期)2014-03-03 02:14:24
