
新生儿先天性高胰岛素血症3 例报告并文献复习
2020-04-04 10:50何艳娟赵明一陈淳媛
临床儿科杂志 2020年3期
蒋 文 何艳娟 赵明一 刘 琳 陈淳媛
中南大学湘雅三医院儿科(湖南长沙 410013)
先天性高胰岛素血症(congenital hyperinsulinemia,CHI)是由于胰岛β细胞不受调控导致胰岛素异常分泌而出现高胰岛素血症的异质性综合征,是新生儿期顽固性低血糖的常见原因之一[1]。CHI是一种常染色体遗传性疾病,发病率为1/50 000~1/30 000[2],其临床表现为反复发作的低血糖,而反复的低血糖可导致神经系统严重受损,在CHI患儿中永久性脑损伤风险可高达25%~50%[3]。目前CHI 的治疗主要分为药物和手术治疗,其中药物治疗以二氮嗪及奥曲肽为主,但CHI 通常对药物具有抗药性,且与CHI 相关基因变异有关,不同基因型对药物治疗反应不同。近年来基因检测在CHI诊断上也起到越来越重要的作用,目前发现有11种基因与CHI发病相关[3]。因此,明确基因突变类型对CHI的诊疗和预后有着重要意义。本文通过对3 例新生儿CHI 临床资料的分析,并复习相关文献,探讨基因检测对CHI 诊断、治疗及预后判断的临床意义。
1 临床资料
例1,男,生后15 分钟出现低血糖,并反复发作。于出生9天入院。患儿系G7P4,胎龄39+4周,经产道分娩,出生体质量2.7 kg,羊水Ⅲ度污染,出生时反应差、呻吟、哭声微弱、全身皮肤青紫,随即送当地医院新生儿科住院治疗,生后15分钟测随机血糖1.6 mmol/L,予以静脉输注葡萄糖治疗后仍有反复低血糖发作。母亲人工流产3 次,母孕期无糖尿病史,父母亲均无糖尿病家族史,父母非近亲结婚,2个姐姐及1哥哥均无低血糖病史。入院体格检查:体温38℃,脉搏114次/min,呼吸48次/ min,体质量2.83 kg。精神、反应差,皮肤、巩膜轻度黄染,无皮疹及皮下出血,呼吸稍促,无吸气三凹征,肺部未闻及干湿啰音,心、腹未见异常,肌张力可,原始反射引出不完全。入院后静脉输注葡萄糖,速度约为10 mg/(kg·min),配合母乳喂养后仍有反复低血糖发作,快速检测血糖波动在2.0~3.8 mmol/L。实验室检查:血清葡萄糖2.0 mmol/L,血清胰岛素13.81 mU/mL;总胆红素156.6 μmol/L,直接胆红素109.4 μmol/L,总胆汁酸94.9 μmol/L;血常规、大小便常规、TORCH、皮质醇、β-羟基丁酸、血氨、乳酸、输血前检查常规、凝血功能均未见明显异常。腹部彩超未见异常。头颅磁共振(MRI)未见明显异常。经医学伦理审核及家长知情同意,采集患儿及其父母EDTA抗凝血各2 mL送至北京康旭医学检验所进行全外显子基因检测。结果显示,患儿ABCC8基因存在c.2517T>A杂合核苷酸变异,该变异导致第839号氨基酸由Ser变为Arg(p.Ser839Arg),为错义变异,受检者父亲该位点为杂合子,母亲该位点未见异常。见图1。患儿予以口服二氮嗪,14 mg/次,q8h;4天后血糖逐渐稳定出院。出院后1个月电话随访,患儿血糖稳定,且已停用二氮嗪。目前患儿1岁3个月,每3个月在当地医院复查1次,血糖未见异常,生长发育与同龄儿相当。
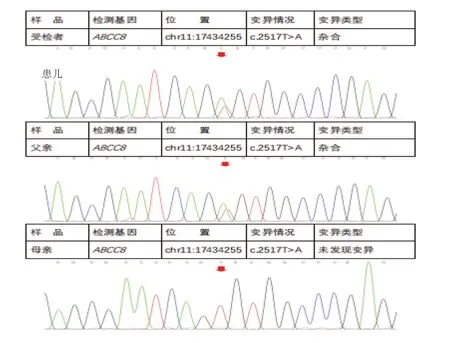
图1 例1 及父母ABCC8 基因测序图
例2,男,生后1 小时出现反复低血糖伴抽搐,于出生6 天入院。患儿系G 2 P 2,胎龄39+6周,剖宫产,出生体质量2.9 kg,羊水Ⅲ度污染。患儿于生后1 小时无明显诱因突然出现抽搐,表现为双眼凝视、四肢抖动、唇周发绀,头部大汗,随即送往当地医院新生儿科就诊。测随机血糖1.2 mmol/L,予以静脉输注葡萄糖治疗后血糖仍难以维持稳定。母孕期无糖尿病病史,父母亲均无糖尿病家族史,父母非近亲结婚,姐姐无生后低血糖病史。入院体格检查:体温37.1℃,脉搏135次/ min,呼吸45次/ min,体质量3.06 kg。精神、反应一般,皮肤红润,无皮疹及皮下出血,呼吸平稳,肺部未闻及干湿啰音,心、腹未见异常,肌张力可,原始反射引出不完全。入院后静脉输注葡萄糖,速度约为10 mg/(kg·min),配合母乳喂养,先后予以氢化可的松及奥曲肽连续静脉滴注后仍有反复低血糖发作,快速检测血糖波动在1.6~4.1 mmol/L。实验室检查:血清葡萄糖1.1 mmol/L,血清胰岛素9.36 mU/mL;总胆红素110.7 μmol/L,直接胆红素16.6 μmol/L,总胆汁酸32 μmol/L;乳酸4.74 mmol/L;皮质醇1.71 μg/dL;血常规、大小便常规、TORCH、β-羟基丁酸、甲状腺功能、血氨、输血前检查常规、凝血功能均未见明显异常。腹部彩超未见异常。头颅MRI示双侧颞叶可见斑片长T1长T2信号影,T2抑水信号稍低。经医学伦理审核及家长知情同意后,采集患儿及父母EDTA抗凝血各2 mL送至北京中同蓝博临床检验所范德瑞尔遗传检测中心进行全外显子基因检测。结果显示,患儿ABCC8基因发现c.3357+71C>T 杂合核苷酸变异,为错义突变,氨基酸改变为p.P149A,受检者父亲该位点未见异常,母亲该位点为杂合子。见图2。患儿予以口服二氮嗪,20 mg/次,q 8 h,5 天后患儿血糖逐渐稳定出院。出院后1 个月电话随访血糖稳定,已停用二氮嗪。目前患儿1岁2个月,定期检测血糖未见异常,生长发育与同龄儿相当。
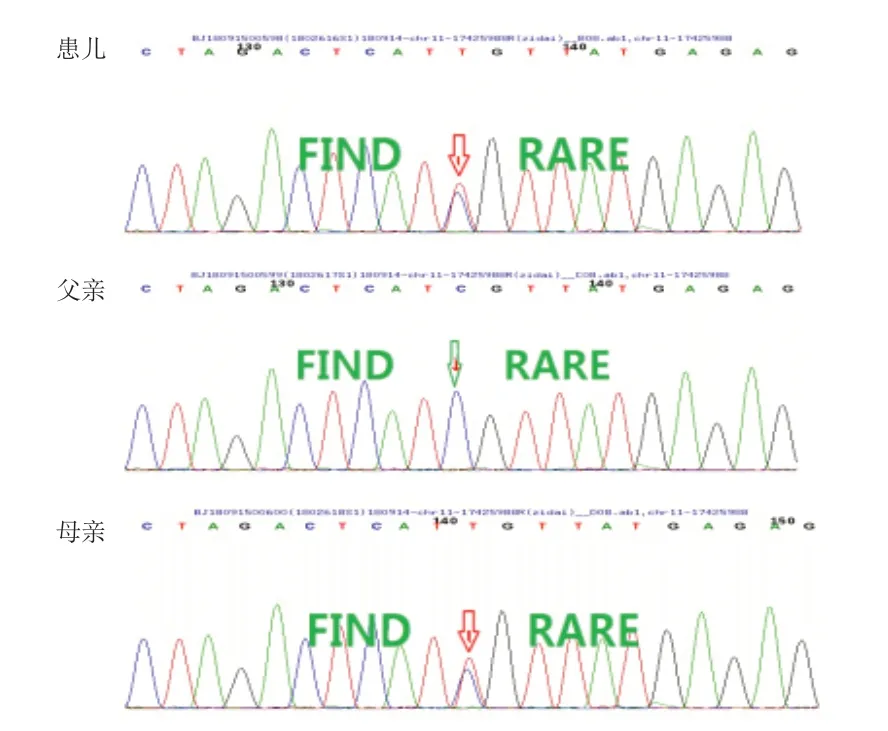
图2 例2 及父母ABCC8 基因测序图
例3,男,生后21分钟出现低血糖并反复发作,于出生19天入院。患儿系G6P2,胎龄36+3周,剖宫产,出生体质量1.89 kg,羊水Ⅲ度污染。因早产及低出生体质量于出生后21分钟入住当地医院新生儿科,快速血糖测定2.2 mmol/L,予静脉输注葡萄糖治疗后患儿血糖仍不稳定。母亲人工流产4 次,母孕期无糖尿病病史,父母亲均无糖尿病家族史,父母非近亲结婚,哥哥无生后低血糖病史。入院体格检查:体温36.8℃,脉搏134次/ min,呼吸50次/ min,体质量2 kg。精神反应欠佳,皮肤欠红润,颜面部及躯干可见少许红疹及红斑,无皮下出血,呼吸稍促,未闻及干湿啰音,心腹未见异常,肌张力可,原始反射引出不完全。入院后静脉输注葡萄糖,速度约为10 mg/(kg·min),配合母乳喂养,先后予以氢化可的松及奥曲肽连续静脉滴注后仍有反复低血糖发作,快速检测血糖波动在0.9~4.8 mmol/L。实验室检查:血清葡萄糖1.2 mmol/L,血清胰岛素17.54 mU/mL;总胆红素28.3 μmol/L,直接胆红素10.5 μmol/L,总胆汁酸59.2 μmol/L;皮质醇63.44 μg/dL;血常规、大小便常规、TORCH、β-羟基丁酸、甲状腺功能、输血前检查常规、凝血功能均未见明显异常。腹部彩超未见异常。头部MRI未见异常。经医学伦理神经及家长知情同意后,采集患儿EDTA抗凝血2 mL送至长沙金域检验所进行KCNJ11基因检测。结果显示,未发现患儿相关基因的异常。因家属拒绝完善全外显子基因检测而未再进一步检测。患儿予以口服二氮嗪,4 mg/次,每8 小时1 次,3天后患儿血糖逐渐稳定出院。出院后1个月电话随访,患儿血糖稳定,仍继续服用二氮嗪,2月龄后逐渐停用二氮嗪。目前患儿1岁半,定期检测血糖未见异常,生长发育与同龄儿相当。
2 讨论
本组3 例CHI 新生儿均在出生后不久出现反复的低血糖发作。因新生儿低血糖的发生较为常见,早期症状无特异性,易被误诊为常见的生后低血糖。但CHI新生儿予以静脉输注较高浓度的葡萄糖,配合母乳喂养,先后予以氢化可的松及奥曲肽连续静脉滴注后,仍可有反复低血糖发作。本组3 例CHI 新生儿实验室检查均提示在低血糖同时出现高胰岛素血症,但血酮体阴性,腹部彩超均未见异常,符合临床CHI诊断。例1及例2经全外显子基因测序发现有ABCC8基因杂合突变,从而进一步确诊CHI,但例3 仅行KCNJ11基因检测,未能从基因上得到进一步的证实。本组3例CHI新生儿经二氮嗪治疗后血糖儿均稳定。
CHI 是一种单基因病,其最佳治疗取决于尽早明确潜在的原因,因此早期遗传诊断具有重要意义。迄今为止已经发现有11 种与CHI 相关的基因,分为两类:与ATP敏感性钾通道(ATP-sensitive potassium channel,KATP)相关的ABCC 8、KCNJ 11基因,与代谢病相关的GLUD1、GCK、HADH、UCP2、SLC16.A1、HNF4A、HNF1A、HK1和PGM1基因[3]。另外还有以综合征形式遗传的高胰岛素血症,如Turner 综合征、Beckwith-Wiedemann综合征和Kabuki综合征等[3]。而不同基因变异可导致其临床表现、对药物的反应及预后有显著的差异。
ABCC8和KCNJ11基因突变是CHI最常见的病因。ABCC8和KCNJ11基因位于染色体 11p15.1上,分别编码SUR1和Kir6.2两个亚单位。胰岛素的释放来源于胰岛β细胞,依赖于细胞膜上的KATP通道,当通道关闭时细胞膜去极化,激活电压门控钙离子通道,胞质内钙离子浓度升高,从而使胰岛素释放入血。KATP通道是由两个亚单位组成的异四聚体,分别是磺酰脲受体-1(sulfonylureas receptor 1 channel,SUR1)亚单位和钾孔(potassium pore,Kir6.2)亚单位[4]。基因变异导致通道改变从而使胰岛β细胞不受调控,最终导致胰岛素的异常分泌。KATP通道相关基因的突变可为隐性或显性遗传,以隐性多见。双等位基因隐性突变可导致质膜KATP通道完全缺乏,而单等位基因的显性突变导致通道功能受损严重降低通道活性,两种突变均表现为弥漫型CHI,对二氮嗪治疗无效。单等位基因隐性突变,只导致胰腺局部区域缺乏质膜KATP通道,表现为局灶型CHI,二氮嗪治疗有效[5]。因此,区分病理类型十分重要,97%的局灶型患儿可以通过手术纠正严重的低血糖,而弥漫型患儿手术治疗无效。
GLUD1基因的显性激活突变造成了高胰岛素血症/高氨血症综合征,这是第二种最常见的CHI 及二氮嗪反应型CHI。GLUD1基因位于染色体10q23.3上,编码谷氨酸脱氢酶(glutamic dehydrogenase,GDH),它是一种线粒体基质酶,表达于肝脏、肾脏、脑及胰岛β细胞。该基因突变时导致三磷酸鸟苷(guanosine triphosphate,GTP)变构抑制敏感性受损,削弱了对GDH 活性的抑制控制,而亮氨酸是GDH 的变构激活剂,二者均使GDH 活性增加,导致谷氨酸氧化增加,临床上表现为空腹和高蛋白饮食后出现低血糖[6]。同时,肾脏中的GDH 过度激活,肾脏中血氨生成增加,导致血氨轻度升高,受影响者可表现为神经系统症状如癫痫、行为障碍和学习障碍等,这可能也反应了大脑中GDH 酶活性的增加[7]。该类型患儿低血糖容易因服用二氮嗪及避免在空腹下摄入蛋白质而得到控制。
GCK基因位于染色体7 p 13 上,编码葡萄糖激酶(glucokinase,GK),其功能是作为胰岛β细胞感受器,调节葡萄糖刺激的胰岛素分泌。GCK基因的显性激活突变降低了胰岛素分泌的葡萄糖阈值,临床上表现为空腹低血糖[8],多见于出生胎龄较大的或体质量较重的婴儿。该类型对二氮嗪反应差,一般需要手术治疗或者连续口服葡萄糖治疗[9]。
短链3-羟酰基CoA 脱氢酶(short-chain,3-hydroxyacyl-coA dehydrogenase,SCHAD)是一种线粒体脂肪酸β氧化酶,由染色体4q25上HADH基因编码,该基因的失活突变是CHI罕见的一种常染色体隐性遗传形式。SCHAD 不仅催化脂肪酸β 氧化,同时对GDH有抑制调节作用[10]。其临床表现与GDH-CHI相似,但无高氨血症及神经系统表现,只有小部分患儿对二氮嗪无反应。
UCP 2基因位于染色体11 q 13.4 上,编码线粒体载体蛋白、解偶联蛋白2(uncoupling protein 2,UCP2),该基因的显性功能缺失突变使UCP2活性降低,促进丙酮酸进入三羧酸循环来增强葡萄糖的氧化,从而导致胰岛素过度释放出现低血糖[11]。该类型对二氮嗪反应敏感。
HNF 4 A和HNF 1 A分别位于染色体20 q 13.2 和12 q 24.31 上,分别编码转录因子肝细胞核因子4 α(hepatocyte nuclear factors 4 α,HNF 4 α)和1 α(HNF1α),这两个基因突变被认为是有关联的,二者调控彼此的表达,形成一个对葡萄糖刺激的胰岛素分泌的重要调节网络[12]。目前发现,该基因突变可致婴儿期高胰岛素血症,也可致成年期糖尿病,目前发病机制仍不明。在婴儿期,低血糖常发生在巨大儿或早期新生儿,严重程度不一,可出现短暂低血糖或持续性高胰岛素血症,二氮嗪治疗有效[13]。
位于染色体1 p 13.2 上的SLC 16.A 1基因编码丙酮酸转运蛋白1(transporter monocarboxylate transporter 1,MCT 1),该基因调控区域的显性突变导致运动后高胰岛素血症。正常情况下胰岛β 细胞表达MCT1,防止丙酮酸及乳酸刺激胰岛素的分泌来调控血糖。SLC16.A1基因启动子突变导致MCT1正常表达抑制丧失,无氧运动时丙酮酸浓度升高,从而导致胰岛素异常分泌导致运动后低血糖[14]。因此,最好的治疗方法就是在剧烈运动期间摄入碳水化合物。
HK1基因位于染色体10q22.1上,编码一种葡萄糖磷酸氧化酶、己糖激酶1(hexokinase 1,HK1),正常情况下HK1抑制低血糖时胰岛素的分泌,而该基因发生突变时,抑制HK1表达,降低胰岛素分泌的葡萄糖阈值,导致高胰岛素血症性低血糖[15]。
PGM1基因位于染色体1p31.3上,编码磷酸葡萄糖醛酸酶-1(phosphoglucomutase 1,PGM1),PGM1催化葡萄糖-6-磷酸酶和葡萄糖-1-磷酸酶可逆相互转化,是糖原形成和降解的关键。该基因隐性失活突变表现为空腹高酮性低血糖,或由于胰岛素对葡萄糖反应过度表现为餐后高胰岛素血症性低血糖[16]。
本组3例CHI新生儿,2例有ABCC8基因杂合突变,其中例1 的突变基因来源于父系,其母亲未携带致病基因,例2的突变基因来源于母系,其父亲未携带致病基因,二者均为杂合突变,结合孟德尔遗传规律,二者均系常染色体隐性遗传。ABCC 8基因与胰岛β 细胞上的KATP通道相关,该基因不同的突变形式导致胰岛β细胞上的KATP通道完全缺乏、活性降低和部分缺乏等不同改变,而不同程度的胰岛β细胞上KATP通道的改变对药物治疗的疗效有显著差异。例1 及例2 患儿的遗传方式为该类型中常见的单基因隐性突变,对二氮嗪治疗有效,患儿均用该药治疗后好转出院。因此,一旦临床考虑CHI后应尽快完善基因检测,早期完善基因检测既能明确诊断,更能指导治疗、评估疗效及预后。
目前CHI的治疗分为药物治疗及手术治疗两个方面。其中药物治疗以二氮嗪及奥曲肽为主,二氮嗪对KATP通道起开放作用,与通道的SUR1亚基结合,使通道开放减少胰岛素释放,因此二氮嗪起作用需要完整的KATP通道,通常二氮嗪对所有KATP通道完整的CHI均有效[17]。奥曲肽是一种生长抑素类似物,通过与生长抑素受体2 和5 结合来抑制胰岛素分泌,同时也可部分抑制KATP通道减少胰岛素分泌[18]。其他药物包括长效生长抑素类似物、硝苯地平及新发现的雷帕霉素、胰高血糖素样肽-1受体拮抗剂等[19]。手术治疗是CHI治疗的另一类重要的治疗方式,组织亚型的分类是手术治疗的关键,目前可通过PET/CT 来识别其病理改变是弥漫型还是局灶型,对于局灶型可行手术切除病灶达到治愈[17]。
综上,CHI 的基因改变多样,不同类型的CHI 治疗方式或药物治疗疗效有显著差异,明确CHI患儿的基因突变类型才能更好地指导治疗,但目前仍有50%的CHI未发现基因突变。因此,积极完善基因检测,建立CHI基因数据库对指导CHI治疗及改善预后具有重要临床意义。
猜你喜欢
保健与生活(2022年9期)2022-05-06
中老年保健(2021年4期)2021-08-22
中医眼耳鼻喉杂志(2019年2期)2019-04-13
科学之谜(2019年3期)2019-03-28
饮食保健(2019年2期)2019-01-17
科学之谜(2018年8期)2018-09-29
妇女之友(2016年11期)2017-01-20
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中华老年多器官疾病杂志(2016年8期)2016-05-14
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
