
Björnstad综合征致病BCS1L基因的新发突变位点研究
2020-04-03 10:39李思源刘婷婷杨淑霞马圣清杨勇
中华皮肤科杂志 2020年2期
李思源 刘婷婷 杨淑霞 马圣清 杨勇
北京大学第一医院皮肤科 北京市皮肤病分子诊断重点实验室 国家皮肤与免疫疾病临床医学研究中心 100034
杨勇现在中国医学科学院 北京协和医学院 皮肤病医院遗传病中心工作,南京210042
Björnstad 综合征(Björnstad syndrome)又名扭曲发综合征,是一种罕见的先天性遗传性疾病,由Björnstad于1965年首次报道,其主要临床特征为先天性头发扭曲和感音性听力丧失。BCS1L为其致病基因,编码一种泛素-细胞色素C还原酶复合体伴侣蛋白,突变后破坏线粒体呼吸小体的组装,降低线粒体电子传递链的活性,增加活性氧的产生,使得对线粒体功能敏感的毛囊和听觉毛细胞产生氧化应激损伤,从而表现为先天性头发扭曲和感音性听力丧失。BCS1L基因突变的病例主要集中在北美和北欧等地区,而国内相关报道较少。我们对收集到的1例Björnstad综合征患者进行BCS1L基因突变研究,以期进一步探究BCS1L基因突变与临床表型之间的联系。
对象与方法
一、对象
患者女,25岁,自出生起即头发稀疏、扭曲且生长速度慢,头发干枯无光泽,易断裂,无头皮瘙痒;眉毛稀,睫毛短(图1)。家长述患者自幼听力正常,无听力障碍,12岁时为改善毛发问题,于后颈部注射不明成分的针剂长达1年,随后突发耳鸣。听力检查示:双耳听力障碍,左耳平均听力85.6 dB,右耳92.9 dB,属于极重度听力障碍,言语识别率0。体检:一般情况良好,身高、体重、智力发育正常。血清微量元素(铜、锌、铁、钙、镁)以及生长激素和性激素检测正常。父母否认近亲婚配,其母妊娠时无特殊疾病和特殊用药史,患者的父母、姐姐及其余家庭成员均未出现头发扭曲或听力障碍等症状。

图1 Björnstad综合征患者临床表现 头发稀疏扭曲,眉毛稀疏。1A:正面照;1B:侧面照
二、方法
1.外周血DNA提取:本研究经北京大学第一医院伦理委员会批准(2016[1024]),患者及家属签署知情同意书后,采集患者及其父母外周静脉血2 ml,用2%乙二胺四乙酸(EDTA)抗凝,常规低渗溶血,酚-氯仿法提取外周血白细胞DNA。
2.聚合酶链反应(PCR)引物设计:根据BCS1L基因序列(来源于ensemble网站,网址:http://asia.ensembl.org/index.html),利用NCBI primer-blast(网址 https://www.ncbi.nlm.nih.gov/tools/primer-blast/)设计7对特异性引物,扩增患者及其父母BCS1L基因全部外显子和侧翼序列,第4号外显子引物序列为:上游5′-CAGTACCCGTACTCAGCACC-3′,下游5′-TAGCCAAAGGGACGCCATTC-3′;第8号外显子引物序列为:上游5′-GCCTCTCTGATGACCGACTC-3′,下游 5′-TGAGCAGTAGCCCACGTACT-3′,引物由北京天一辉远生物科技有限公司合成。
3.PCR反应体系及条件:以PCR DNA扩增仪(Eppendorf,德国)进行PCR反应,反应体系25 μl:2 × KAPA2G Fast Genotying Mix 12.5 μl,上下游引物各1.0 μl(10 pmol/μl),DNA 1 μl(约200 ng),补充ddH2O至25 μl;扩增条件:94 ℃预变性5 min;94℃变性30 s,60℃退火30 s,72℃适温延伸45 s,共30个循环;最后72℃延伸10 min。1.5%琼脂糖凝胶电泳检测PCR产物,所有PCR产物经纯化后送至北京天一辉远生物科技有限公司,包括目的外显子所有序列以及两侧至少30 bp侧翼序列,经ABI3730xl全自动DNA测序仪测序,结果与Ensemble genome browse所公布的序列双向对比,使用BioEdit sequence Alignment Editor软件进行分析,并通过预测软件(http://www.mutationtaster.org/)对突变位点进行预测。
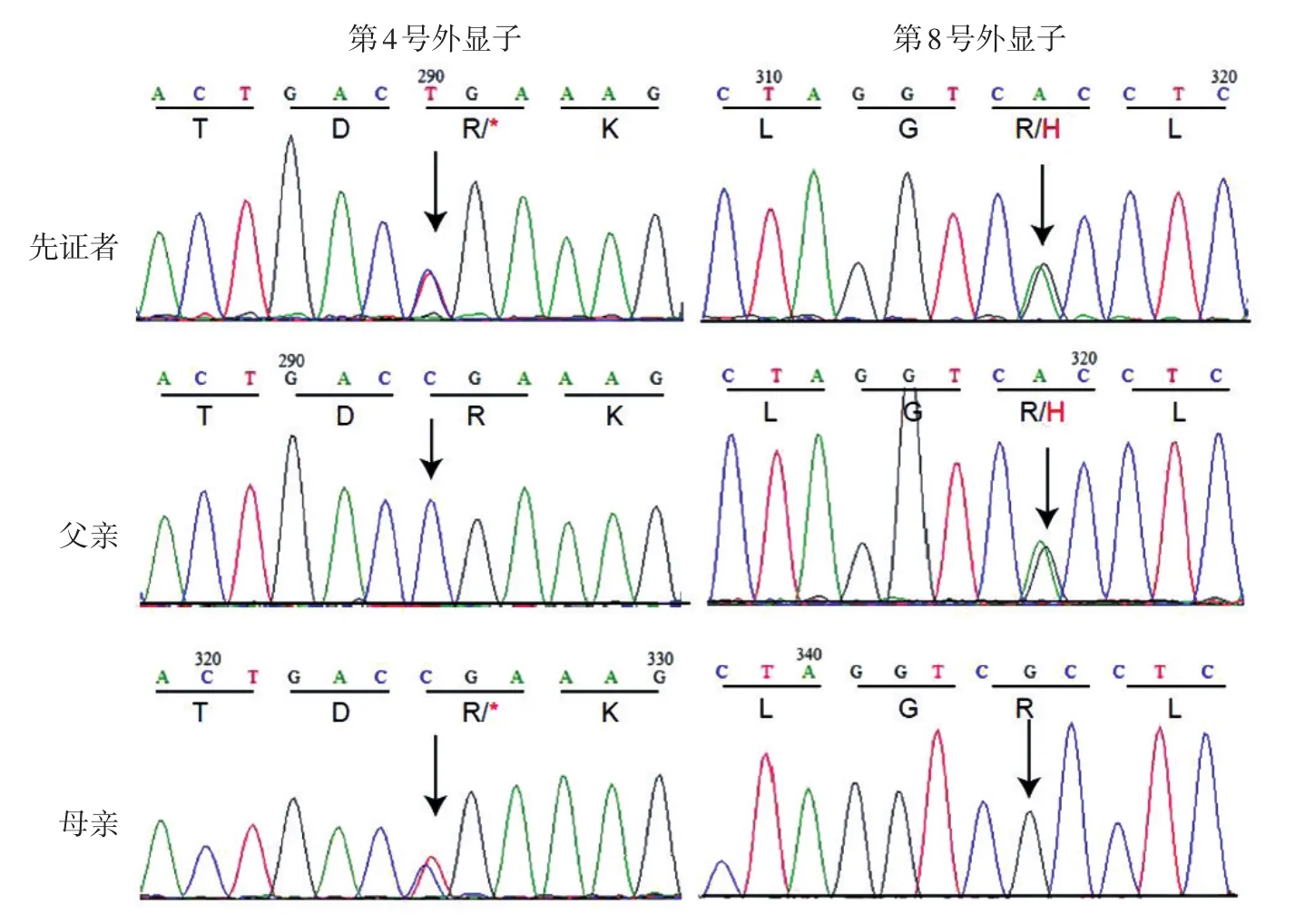
图2 Björnstad综合征先证者及其父母的BCS1L基因突变分析 先证者BCS1L基因第4号外显子发生c.430 C>T杂合突变(p.R144*),第8号外显子发生c.917 G>A杂合突变(p.R306H);先证者父亲BCS1L基因第4号外显子正常,第8号外显子发生c.917 G>A杂合突变(p.R306H);先证者母亲的BCS1L基因第4号外显子发生c.430 C>T杂合突变(p.R144*),第8号外显子正常
4.电镜检查:用细镊子轻取患者头发若干根,送至电镜室进行样本处理并使用扫描电镜(AmRay 1000A)检查。
结 果
一、PCR扩增结果
以该患者及其父母的DNA为模板,7对引物均可扩增出各自产物,长度为436~714 bp。
二、DNA测序结果
患者BCS1L基因第4号外显子上存在杂合无义突变(图2):第430位碱基发生C→T(c.C430T)杂合突变,使第144位密码子CGA→TGA,导致其编码氨基酸序列由精氨酸突变为终止密码子(p.R144*),通过预测软件(http://www.mutationtaster.org/)进行预测,显示出现无义介导的mRNA降解而致病,该预测结果高度可信(得分为1);第8号外显子上存在杂合错义突变(图2):第917位碱基发生G→A(c.G917A)杂合突变,使第 306位密码子CGC→CAC,导致其编码氨基酸序列由精氨酸突变为组氨酸(p.R306H)。对先证者父母BCS1L基因进行测序,结果显示,其父母均为携带者:患者父亲的BCS1L基因第8号外显子发生c.917 G>A杂合突变(p.R306H),第4号外显子正常(图2);患者母亲的BCS1L基因在第4号外显子发生c.430 C>T杂合突变(p.R144*),第8号外显子正常(图2)。突变均由反向测序得到验证。
三、电镜检查结果
扫描电镜下见患者头发以不规则的间隔出现扁平、沟槽和沿长轴扭曲,发干表面毛小皮边缘不清、粗糙,伴部分翘起(图3)。
讨 论
Björnstad综合征为常染色体隐性遗传,患者通常出生后不久即出现头发异常,包括头发扭曲,无规则扁平,生长速度慢,发质干枯无光泽,易断裂,随着年龄增长而加重,眉毛、睫毛亦可受累。电镜下发干以不规则的间隔出现沟槽,绕轴旋转,从而引起毛发的自发性断裂,发干表面毛小皮边缘不清、粗糙,伴部分翘起。

图3 Björnstad综合征先证者毛发扫描电镜图像 3A:头发出现扁平、凹槽和纵嵴,毛小皮边缘不清、粗糙,伴部分翘起(×500);3B:头发出现沿发干长轴的扭曲(×500)
扭曲发的形成可以是先天性的,也可以继发于各种原因的斑秃而引起[1]。先天性扭曲发亦可见于各种罕见的综合征,如:Crandall综合征[2]、Menkes综合征[3]、外胚层发育不良、神经缺陷和代谢紊乱等,可通过基因测序,检测血清铜、生长激素、性激素水平等方法鉴别。
1965年,Björnstad首先将先天性头发扭曲和感音性听力丧失作为一组遗传表型,报告了8例扭曲发患者,其中5例并发感音性听力障碍,并建议扭曲发的低龄儿童应尽早进行听力测试[4]。1998年,Lubianca Neto等[1]将 Björnstad 综合征的致病基因定位于染色体2q34-36。2007年,该课题组研究发现,BCS1L基因编码一种泛素-细胞色素C还原酶复合体伴侣蛋白,位于线粒体内膜上,在线粒体呼吸链组装中帮助Fe/S蛋白插入到复合体Ⅲ的前体中,是线粒体呼吸链复合体组装所必需的伴侣蛋白[5]。BCS1L蛋白包含两个功能域:促进线粒体内膜的易位及插入区域(1~89个氨基酸)和ATP活动相关的ATP酶家族的区域(220~399个氨基酸)。而突变基因编码形成的BCS1L蛋白改变了蛋白之间的相互作用,增加了缺乏Fe/S蛋白插入的线粒体呼吸链复合体Ⅲ前体的积累,破坏了线粒体呼吸小体(人类线粒体呼吸作用的基本单位)的组装,降低了线粒体电子传递链的活性,增加了活性氧的产生。
BCS1L基因突变导致的Björnstad综合征显示了耳朵和头发组织对线粒体功能的敏感性,特别是对活性氧产生的敏感性,快速分裂的毛囊细胞较容易受到活性氧水平增加的影响,而已有模型支持活性氧水平对耳毒性的作用,如耳毒性抗生素对听觉毛细胞造成的氧化应激损伤[6]等。除了仅有耳朵和毛发受累的Björnstad综合征之外,BCS1L基因突变造成的线粒体呼吸链复合体Ⅲ的缺乏,也会导致其他广泛的疾病表型:如神经系统疾病[7],局灶性癫痫、肌无力和视网膜萎缩,以及更为严重的线粒体复合体Ⅲ缺乏症[8](出生时出现代谢性酸中毒、新生儿近端肾小管病变、肝脏损害和神经受累)和Gracile综合征[9](表现为生长迟缓、氨基酸尿、胆汁淤积、铁超载和乳酸中毒以及早夭)等,后两者为严重的线粒体疾病,是新生儿致命性疾病。
从基因学角度分析,所有BCS1L基因突变均破坏了线粒体呼吸小体的组装,降低了线粒体电子传递链的活性,并且增加了活性氧的产生。而活性氧的产生情况与BCS1L基因突变的临床表型严重程度相关。此外,BCS1L基因突变造成的表型差异也可能与线粒体的异质性、组织对能量需求的可变性以及组织对活性氧的敏感性有关。国外蛋白组学研究显示,BCS1L蛋白存在p.I106*、p.R114W、p.R183H、p.A273fs*27、p.R291*、p.Q302E、p.R306H等改变的病人仅出现典型的Björnstad综合征特征——出生时即表现出毛发异常以及先天性听力障碍,无其他疾病表现;当BCS1L蛋白存在p.G35R和 p.R184C[5]突变时,患者除了具有典型的Björnstad综合征特征外,还存在如生长迟缓、发育迟缓和肌张力减退等轻度线粒体复合体Ⅲ缺乏症的表现;当患者BCS1L蛋白存在p.R56*、p.S78G、p.P99L、p.R144Q、p.R155P、p.S277N、p.V327A、p.V353M等突变时,表现出了更为严重的线粒体复合体Ⅲ缺乏症和Gracile综合征的临床表型。患者复杂的多系统临床表现以及早夭限制了对BCS1L基因不同突变位点及其对应临床表型的研究。国内报道的BCS1L基因突变致病的病例[10]较少,且均为Björnstad综合征,突变位点包括p.R306C和p.R186*[11]、p.A273fs*27 和 p.R306H[12]等,而线粒体复合体Ⅲ缺乏症和Gracile综合征尚未见报道。
本文报告的患者自幼毛发稀疏、扭曲,但听力正常;其听力障碍出现时间较晚,并且有药物诱发因素存在,而又区别于药物中毒性耳聋的听觉系统慢性中毒(常出现于治疗后1~2周,逐渐加重)及可能的前庭系统受累(可出现眩晕和平衡失调)症状,本例患者的听力障碍发生于持续用药1年后,无渐进性加重过程,无前庭系统受累表现。对本例患者及其父母的BCS1L基因进行测序,结果显示,患者第4号外显子发生c.430 C>T杂合突变(p.R144*),其母亲为该突变的携带者;患者第8号外显子发生c.917 G>A杂合突变(p.R306H),其父亲为该突变的携带者。患者BCS1L基因第4号外显子c.430 C>T杂合突变(p.R144*)为该基因突变导致Björnstad综合征的新发致病突变位点,既往虽有第144位氨基酸由精氨酸突变为谷氨酰胺(p.R144Q)的报道[5],但突变为终止密码子的报告尚属国际首例,通过预测软件(http://www.mutationtaster.org/)预测,显示BCS1L基因第4号外显子发生c.430 C>T杂合突变后出现无义介导的mRNA降解,该预测结果高度可信(得分为1),推测提前终止的BCS1L蛋白出现了功能缺失而致病;第8号外显子上的突变位点位于BCS1L蛋白的ATP活动相关的ATP酶家族功能域的外表面,既往文献已报道该点突变影响了BCS1L蛋白与其他蛋白的相互作用,进而影响线粒体呼吸链ATP依赖的组装过程,从而导致疾病的发生[5]。结合患者的毛发异常及其BCS1L基因的突变情况,诊断为Björnstad综合征,并对其不典型的听力障碍进行报告。目前本例患者仍在随访中。
综上所述,本文在既往研究的基础上,首次报道BCS1L基因第144位密码子CGA→TGA导致的编码终止为该基因突变导致Björnstad综合征的新发致病突变位点,为进一步明确Björnstad综合征基因型和表型的关系积累了新证据。随着分子遗传学的进展,可以通过基因诊断,对患者进行预后评估,也可以通过产前诊断,阻止疾病在家族中的延续。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
现代农业科技(2022年5期)2022-12-14
农业工程学报(2022年13期)2022-10-09
科学导报(2022年11期)2022-03-03
作物学报(2022年3期)2022-01-22
种子(2021年3期)2021-04-12
智慧健康(2020年9期)2020-12-03
中华养生保健(2020年5期)2020-11-16
中华肩肘外科电子杂志(2019年4期)2019-08-24
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
