
高效液相色谱法定量分析奥曲肽
2020-03-26 06:37向童欣黄永鹏唐慧钟辉陈博
化学分析计量 2020年2期
向童欣,黄永鹏,唐慧,钟辉,陈博
(军事科学院防化研究院,北京 102205)
奥曲肽(Octreotide,OCT)是由八个氨基酸组成的环状多肽化合物,化学名称为D-苯丙胺酰-L-半胱氨酰-L-苯丙胺酰-D-色氨酰-L-赖氨酰-L-苏氨酰-L-半胱氨酰-L-苏氨醇(2 →7)环二硫化物,化学结构式如图1 所示。OCT 是首例获批上市的人工合成生长抑素类药物,与天然生长抑素(Somatostatin, SST)相比,具有更强的作用效果与更长的半衰期[1],对腺体分泌(生长激素、促甲状腺激素、胰岛素、胰高血糖素、降钙素等)、内脏血流、平滑肌收缩、细胞增殖、肿瘤增长等具有抑制作用,是目前临床上治疗肢端肥大症的首选药物[2],同时在治疗急慢性胰腺炎[3–4]、上消化道出血[5–6]、消化道内分泌/非内分泌性肿瘤[7]、肺癌[8]等领域应用广泛。近年来,OCT 原料药合成、新剂型开发、药理学等研究备受关注,而上述研究均需要以OCT 的定量分析作为依据。
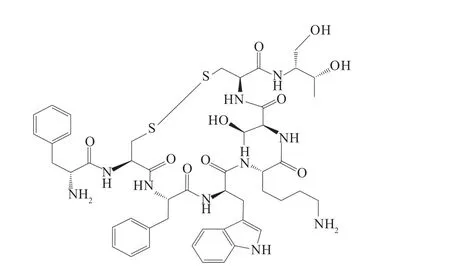
图1 奥曲肽的化学结构式
目前OCT 定量分析方法主要有高效液相色谱(HPLC)法[9–14]、高效液相色谱–质谱(HPLC–MS)法[15–16]、酶联免疫吸附测定(ELISA)法[17]、放射性免疫分析(RIA)法[18]、共振瑞利散射(RRS)法[19]等。其中,HPLC 法设备费用相对较低,操作较简单,在OCT 原料药及其制剂纯度与杂质分析、载药量、稳定性、质量一致性、体外溶出度等评价中应用最为广泛。目前文献报道的关于奥曲肽的HPLC定量分析方法主要是参照《中国药典》中醋酸奥曲肽(Octreotide Acetate,OA)测定含量的方法[20],即采用不同浓度的四甲基氢氧化铵水溶液和不同体积的乙腈分别作为流动相A 和流动相B,然后进行梯度洗脱。该方法存在流动相配制繁琐、分析时间长(大于30 min)、压力不稳定、基线噪声和漂移大影响分析精密度和稳定性、色谱柱需要再生处理、磷酸盐容易析出损坏色谱柱与柱塞泵等不足。笔者采用乙腈–固定浓度的高氯酸水溶液体系等度洗脱,分别考察了流动相pH 值和配比对OCT 色谱行为的影响,优化相关色谱条件,建立了新的HPLC 定量分析OCT 的方法,该方法操作简便,测定快速,结果准确,适用于OCT 原料药与制剂的分析评价。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Agilent 1260 型,美国安捷伦科技有限公司;
电子分析天平:XP105DR 型,上海梅特勒–托利多仪器有限公司;
酸度计:Sevenmulti 型,上海梅特勒–托利多仪器有限公司;
奥曲肽标准品:纯度为99.1%,批号为2018053003–1,浙江湃肽生物有限公司;
奥曲肽原料药样品:实验室自制;
乙腈:色谱纯,美国默克公司;
高氯酸:分析纯,北京百灵威公司;
实验用水为娃哈哈纯净水。
1.2 色谱条件
色 谱 柱:Eclipse plus C18(4.6 mm×250 mm,5 μm,美国安捷伦科技有限公司);流动相:乙腈–0.25%高氯酸水溶液(体积比为30∶70);检测波长:210 nm;流量:1 mL/min;柱温:25℃;进样体积:5 μL。
1.3 溶液配制
OCT 储备溶液:219.0 μg/mL,精确称取21.9 mg 奥曲肽标准品,置于100 mL 容量瓶中,加入适量纯水溶解并定容至标线,摇匀。
系列OCT 标准工作溶液:分别精密移取0.2,0.5,1.0,2.0,4.0,6.0,10.0 mL 的OCT 储 备 溶 液 至10 mL 容量瓶中,加入纯水稀释并定容至标线,摇匀,配制成奥曲肽的质量浓度分别为4.38,10.95,21.90,43.80,87.60,131.40,219.00 μg/mL 的 系 列OCT 标准工作溶液。
1.4 样品处理
精确称取20 mg 奥曲肽原料药样品,置于250 mL 容量瓶中,加入适量纯水溶解,超声5 min,冷却后用纯水定容至标线,摇匀,静置后用0.45 μm 微孔滤膜过滤,滤液即为待测样品溶液。
2 结果与讨论
2.1 流动相pH 值确定
在乙腈–高氯酸水溶液体积比为30∶70 条件下,通过改变高氯酸水溶液中高氯酸含量,配制pH 0.6 至pH 6.4 的系列流动相体系,考察流动相pH 值对OCT 色谱行为的影响,结果见表1。

表1 流动相pH 值对奥曲肽色谱行为的影响
由表1 可知,当流动相pH ≥2.24 时,OCT 主峰与溶剂峰不能完全分离;当流动性pH 值≤1.94时,OCT 主峰与溶剂峰完全分离,随流动相pH 值的减小,OCT 的保留时间逐渐延长,色谱峰的对称性和峰宽逐渐增加,峰高逐渐减小,峰面积无显著差异。当高氯酸含量为0.25%,即流动相体系的pH值为1.7 时,pH 对OCT 色谱行为影响较小,OCT主峰保留时间小于9 min,且峰形尖锐、对称性良好。综合保留时间、峰形对称性、峰高、峰宽等因素选择在高氯酸水溶液中加入0.25%高氯酸,即流动相pH值为1.7。
2.2 流动相配比对选择
分别以体积比为10∶90,20∶80,25∶75,30∶70,35∶65,40∶60 的乙腈–0.25%高氯酸水溶液作为流动相进样分析,考察不同流动相配比对OCT 色谱行为的影响,上述流动相体系的pH 值均在1.70±0.05之间,在此范围内pH 对OCT 色谱行为影响较小,实验结果如图2 所示。
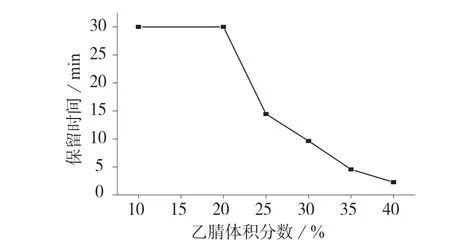
图2 流动相中乙腈体积分数对奥曲肽保留时间的影响
由图2 可以看出,随着流动相体系中乙腈体积分数的增大,OCT 的保留时间随之减小。当乙腈与0.25%高氯酸水溶液体积比为10∶90 和20∶80 时,流动相洗脱能力不够,30 min 内未出峰。当二者的体 积 比 为25∶75,30∶70,35∶65 时,OCT 主 峰 与溶剂峰完全分离,保留时间分别为14.45,8.92,4.56 min,其中,当二者的体积比为30∶70 时,色谱峰峰形尖锐,且对称性最好。当二者的体积比为40∶60时,OCT 主峰与溶剂峰不能完全分离。综合考虑,选择体积比为30∶70 的乙腈– 0.25%高氯酸水溶液作为流动相,OCT 的保留时间为8.92 min,色谱图如图3 所示。
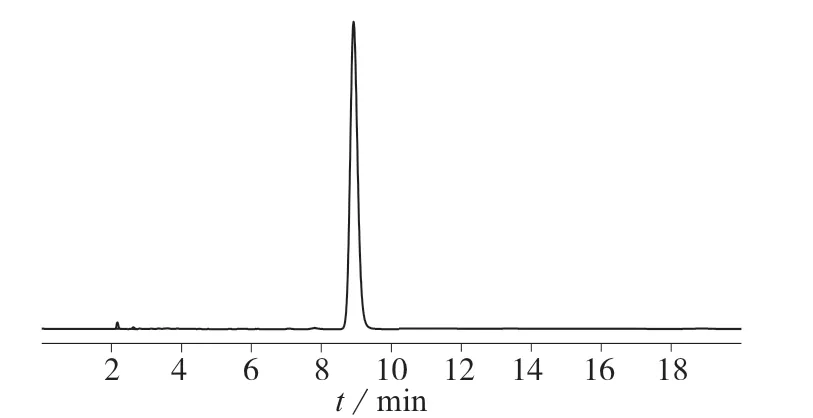
图3 OCT 的色谱图
2.3 线性方程与检测限
将系列OCT 标准工作溶液用0.45 μm 微孔滤膜过滤,在1.2 色谱条件下分别进行测定,每一浓度的OCT 标准工作溶液重复进样2 次,以两次测定值的平均值作为色谱峰面积的测定结果。以OCT 的质量浓度(X)为横坐标,以色谱峰面积(Y)为纵坐标,绘制标准工作曲线,计算得线性方程为Y=11.579X–31.461,相关系数为0.999 9。表明OCT的质量浓度在4.38~219 μg/mL 范围内线性关系良好。
将质量浓度为4.38 μg/mL 的OCT 标准工作溶液逐步稀释,在1.2 色谱条件下分别进样测定,计算相应色谱峰高和平均基线噪音之比。以3 倍信噪比(S/N=3)时的浓度为最低检测限浓度,以10 倍信噪比(S/N=10)时的浓度为最低定量限浓度,将检测限浓度乘以进样体积5 μL,得最低定性与定量检测限分别为1.10,2.19 ng。
2.4 精密度试验
按1.4 方法分别对5 份奥曲肽原料药样品进行处理,得到5 组不同质量浓度的OCT 样品溶液,在1.2 色谱条件下分别连续进样测定5 次,结果见表2。
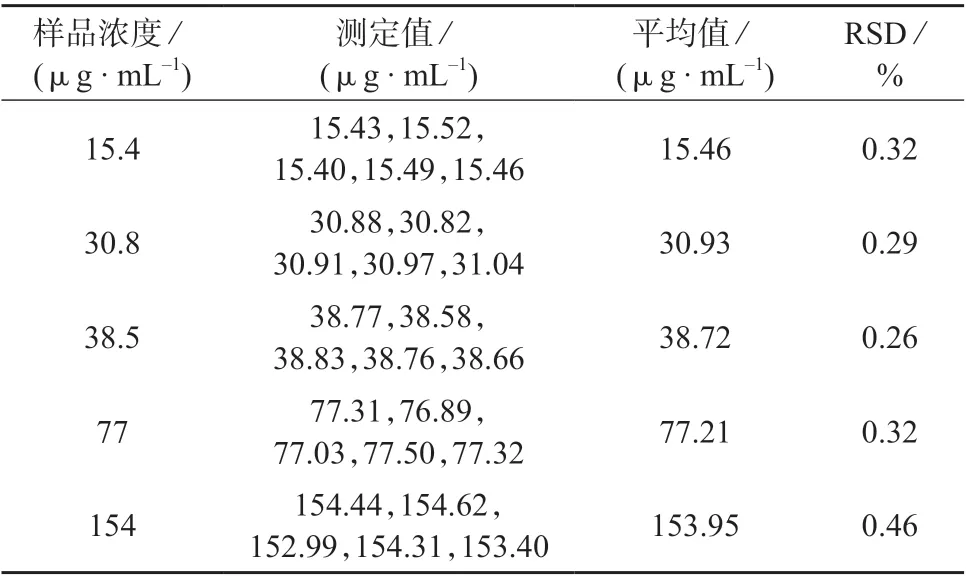
表2 精密度试验结果
由表2 可知,5 组不同质量浓度的OCT 样品测定结果的相对标准偏差为0.26%~0.46%,表明该方法精密度良好。
2.5 加标回收试验
分别将含有180,300,600 μg 奥曲肽的溶液样品添加到空白样品基质(纯水)中,定容配制成低、中、高3 种加标浓度的样品,经0.45 μm 微孔滤膜过滤后,在1.2 色谱条件下进行测定,每个浓度样品平行测定3 次,根据色谱峰面积及线性方程计算回收率,结果见表3。由表3 可知,样品加标回收率为97.41%~100.26%,表明该方法具有较高的准确度。

表3 加标回收试验结果
2.6 稳定性试验
选取质量浓度分别为10.95,43.80,219.00 μg/mL 的OCT 标准工作溶液,在1.2 色谱条件下,分别通过在一天内每隔2 h 测定一次、共计5 次和每天测定一次、连续测定5 天的方式,考察分析方法的日内与日间稳定性,结果见表4。日内测定结果的相对标准偏差为0.32%~0.37%,日间测定结果的相对标准偏差为0.47%~1.31%.表明该方法具有较高的稳定性。

表4 日内与日间稳定性试验结果
2.7 实际样品测定
按1.4 方法分别对实验室制备的5 个批次的奥曲肽原料药样品进行处理,在1.2 色谱条件下,对每个样品平行测定3 次,取平均值作为测定结果,根据色谱峰面积及线性方程计算得5 个批次原料药样品中奥曲肽含量分别为98.3%,98.2%,98.0%,98.7%,98.2%。表明所制备的原料药具有较高的纯度与良好的批次稳定性。
3 结语
建立了以乙腈–高氯酸水溶液为流动相体系的奥曲肽等度洗脱HPLC 定量分析方法,着重考察了流动相pH 值及配比对OCT 色谱行为的影响,与常规药典方法相比,避免了含有磷酸的复杂流动相体系以及梯度洗脱存在的不足。该方法操作简便,检测快速,结果准确,可用于OCT 原料药与制剂的分析评价,以促进OCT 的药学研究与临床应用。
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
中国经济周刊(2021年22期)2021-12-07
医学食疗与健康(2021年27期)2021-05-13
智慧健康(2021年33期)2021-03-16
中国药学药品知识仓库(2021年18期)2021-02-28
环球时报(2020-02-20)2020-02-20
传媒评论(2019年6期)2019-10-14
科学与财富(2018年26期)2018-10-24
中国科技纵横(2018年10期)2018-07-27
中国经济信息(2017年17期)2017-09-09
