
ACE2小干扰RNA对平滑肌细胞AT1受体蛋白表达及其下游信号通路的影响
2020-03-20 00:25龚晶婧李荣通卢卓强许昌声王华军晋学庆
中国药理学通报 2020年3期
龚晶婧,李荣通,卢卓强,许昌声,王华军,晋学庆
(1.福建医科大学附属第一医院心内科,2.福建省高血压研究所,3.福建医科大学附属第一医院干部病房,福州 350004)
肾素-血管紧张素系统(renin-angiotensinsystem,RAS)是人体内重要的内分泌系统,在调节机体水电解质平衡、血压稳定及心肾功能方面起着举足轻重的作用,其成员主要包括肾素、血管紧张素原、血管紧张素转换酶以及血管紧张素Ⅰ、 Ⅱ、Ⅲ、Ⅳ等。其中血管紧张素Ⅱ(angiotensionⅡ,AngⅡ)为该系统发挥上述作用的重要效应分子,在平滑肌细胞(vascular smooth muscle cells,VSMCs)上与其l型受体(AngⅡ type 1 receptor,ATlR)结合后,经丝裂原激动蛋白激酶(mitogen activated protein kinases,MAPK)等多条信号通路后启动VSMCs的增殖、细胞肥大等,最终导致血管收缩、血压升高及心肌肥厚等心血管病理改变。[1]
2000年,Donoghue等和Tipnis等几乎同时分别从人淋巴瘤和扩心病心衰患者左心室组织的cDNA文库中克隆出与人类血管紧张素转化酶(angiotensin converting enzyme,ACE)相关的羧肽酶[2],称之为ACE2。目前人类ACE2基因已被定位于X染色体Xp22位点上,基因全长40kb,含有18个外显子,其中17个与ACE外显子大小、结构、形态相似,表明二者来自同一祖先基因[2]。此外,与ACE的全身广泛分布不同,ACE2在器官水平上主要分布于心脏、肾脏及睾丸,细胞水平上主要分布于心脏、肾脏血管的内皮细胞[2],此外还在胃肠道、脑、肺里有少量分布。 ACE2催化产物Ang-(1-7)与其特异性受体Mas结合后具有降低血压、调节机体水盐平衡、抗心肌肥厚、逆转心室重构等重要心血管保护功能[3],因此ACE2主要作用是一方面增加AngⅡ的降解而减少AngⅡ导致的细胞增殖、肥大、血管收缩等不利效应,另一方面通过生成Ang(1-7)与其特异受体Mas结合舒张血管、抑制细胞增殖而发挥拮抗AngⅡ的作用。由此发现RAS另一分支:ACE2-Ang(1-7)-Mas轴,且其与传统的ACE-AngⅡ-ATlR轴二者相互拮抗、平衡,对维持血压和心血管正常结构及功能有重要意义。
RNA干扰(RNA interference,RNAi)为一种由双链RNA(double-stranded RNA,dsRNA)诱发的基因沉默现象,与dsRNA具有同源序列的mRNA被降解,从而达到抑制该基因的表达。是生物体进化过程中由于基因重复序列、基因突变及病毒感染等所引起基因组不稳定的保护机制。对RNA干扰现象的最初认识是1998年Fire和Blello用线虫和果蝇实验所得[4]。RNAi具有高度特异性、高效性、遗传性、高度稳定性等特点。目前,随着对RNAi技术研究的深入,表明其在基因功能、基因表达、基因治疗以及药物筛选等领域有着广阔的应用前景。因此,本研究运用RNAi技术,探讨ACE2蛋白表达变化对ATl受体、ERK1/2蛋白、STAT3蛋白磷酸化水平的影响及其可能机制,初步探讨ACE2拮抗AngⅡ的生物学作用机制,为探索心血管疾病治疗机制尽可能开辟新的研究领域。
1 材料与方法
1.1 主要试剂和试剂盒细胞培养液DMEM和胎牛血清购自Invitrogen公司,AngⅡ、ACE2 siRNA购自Sigma公司(美国),兔抗大鼠ACE2一抗、兔抗大鼠ATlR一抗(英国Abcam公司)。兔抗大鼠p-ERK1/2、ERK1/2、p-STAT3、STAT3一抗(美国CST公司),小鼠β-actin一抗、羊抗大鼠和小鼠二抗(美国Santa Cruz公司)。DMEM培养基、胎牛血清(美国Gibco公司)。
1.2 实验动物及慢病毒体质量为(120~130) g普通成年SD ♂大鼠购自上海斯莱克实验动物有限公司,动物使用许可证号:SCXK(沪)2007-0005。慢病毒重组ACE2表达载体购自上海吉凯因化学技术有限公司。
1.3 仪器低温高速离心机(德国Sigma公司),垂直电泳仪(美国BioRad公司)。
1.4 SD大鼠胸主动脉VSMC的培养SD大鼠胸主动脉VSMC采用组织贴块法培养。
1.5 实验分组① 空白对照组; ② GFP组; ③ 脂质体组; ④ ACE2组; ⑤ si-ACE2组; ⑥ AngⅡ 组; ⑦ AngⅡ+ACE2组; ⑧ AngⅡ+ACE2+si-ACE2组; ⑨ AngⅡ+si-ACE2组; ⑩ scrambled si-RNA组。
1.6 si-ACE2的制备ACE2的小干扰RNA(si-ACE2)由Sigma公司合成,与ACE2 mRNA碱基序列配对,G/C含量在30%~70%之间,并在每条单链3′端外带两个T碱基,再与编码链形成双链。用0.1%的DEPC水稀释至浓度20 μmol·L-1,-20℃保存。ACE2小干扰RNA的碱基序列:5′-GATAACTTAGTCCCCTCCTTCCTT-3′,5′-GGAAGGAGGGGACUAAGUUAUCTT-3′,si-ACE2各组,按如下步骤准备si-RNA-lipo2000混合液(试剂的用量和体积参照小干扰RNA说明书):① 稀释转染试剂:lipofectamine2000(简称lipo2000): 取不含胎牛血清的DMEM培养基100 μL稀释2 μL lipo2000,轻轻混合,室温孵育5 min;② 稀释si-RNA:取不含胎牛血清的DMEM培养基100 μL稀释2.5 μL siRNA(终浓度50 nmol·L-1),轻轻混合;③ 稀释好的lipo-2000经过孵育后与上述混好的si-RNA轻轻摇晃,使之混匀,室温孵育20~30 min。
1.7 细胞的接种、转染、干预取第三代平滑肌细胞按1×105/孔种植于12孔板,并加入1 mL含20%胎牛血清DMEM培养基,直至孔板内细胞长满50%左右,将所有细胞随机分组(分组方法详见实验分组)。Lentiviral-GFP组感染条件:5 μL(1 mg·mL-1) polyprene,975 μL 20%完全培养液DMEM,20 μL (1×108TU·mL-1)病毒液; Lentiviral-ACE2组各组感染条件:5 μL (1 mg·mL-1) polyprene,975 μL 20%完全培养液DMEM, 20 μL (1×108TU·mL-1) 病毒液;各si-ACE2组加入上述制备好的siACE2-lipo2000混合液,轻轻摇晃使之混匀;脂质体组仅加入lipo2000;scrambled siRNA组转染步骤同各siACE2组。各组感染终体积均为1 mL。Lentiviral-ACE2转染96 h,AngⅡ干预浓度为10-7mol·L-1,干预8 h,培养72 h。
1.8 Western blot测定各组ACE2、AT1R蛋白表达、ERK1/2蛋白和STAT3蛋白磷酸化水平将上述各组干预培养72 h后,将培养液倒掉,在每孔中加入200 μL RIPA(组成:50 mmol·L-1Tris-HCl pH 7.4,150 mmol·L-1NaCl,1%NP-40,0.1% SDS),提取蛋白。10%的SDS-PAGE电泳、转膜,5%的脱脂奶粉室温下封闭1 h,加入兔抗大鼠ACE2(1 ∶500),兔抗大鼠ATIR(1 ∶800),兔抗大鼠p-STAT3、STAT3(1 ∶800),4 ℃过夜,用TBST(组成:1 mol·L-1Tris-HCl 10 mL pH 7.4,NaCl 8.8 g,吐温20 0.05%)洗3次,每次10 min。加入二抗(1 ∶1 500),室温培育1 h,TBST洗3次,每次10 min。显色、曝光,经Guantity one软件分析吸光度值(A),并与内参照的比值作为半定量指标进行比较。
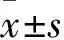
2 结果
2.1 Western blot检测ACE2的siRNA干预Lentiviral-ACE2转染的SD大鼠VSMCs后各组ACE2的蛋白表达各组细胞培养72 h后,提取细胞ACE2蛋白,行Western blot检测各组ACE2蛋白表达量,从Fig 1A可看出:空白对照组、GFP组及脂质体组ACE2蛋白有少量表达,且ACE2组ACE2蛋白表达量为各组中最高,较上述各组明显升高,差别具有统计学意义,说明病毒载体构建及转染血管平滑肌细胞成功及其转染后能明显提高ACE2蛋白的表达; si-ACE2组ACE2蛋白表达水平则明显低于空白对照组、GFP组和脂质体组,差异具有统计学意义,表明siRNA能明显抑制ACE2蛋白的表达;AngⅡ组ACE2蛋白表达量较空白对照组、GFP组及脂质体组亦明显减少,提示AngⅡ能抑制ACE2蛋白的表达;AngⅡ+si-ACE2组的ACE2蛋白表达水平则明显低于AngⅡ组;AngⅡ+ACE2+si-ACE2组的ACE2蛋白表达水平则低于AngⅡ+ACE2组,说明siRNA与AngⅡ两者对ACE2蛋白表达有协同抑制作用。以上各组结果说明各si-ACE2干预组ACE2蛋白表达量较相对应的无si-ACE2干预组明显减少,表明si-ACE2能明显抑制ACE2蛋白的表达,且si-ACE2与AngⅡ对ACE2蛋白表达的抑制具有协同作用。

Fig 1 The ACE2 (A) and AT1R (B) protein expression after ACE2 si-RNA interference in VSMCs measured
1. Control group; 2. Lentiviral-GFP group (GFP);3. Lipofectamine 2000 group;4. Lentiviral-ACE2 group;5. si-ACE2 group;6. AngⅡ group;7. AngⅡ+lentiviral-ACE2 group; 8. AngⅡ+lentiviral-ACE2+si-ACE2 group; 9. AngⅡ+siACE-2 group; 10. Scrambled siRNA group.*P<0.05vscontrol,GFP and lipofectamine 2000 group;#P<0.05vsAngⅡ group;##P<0.05vsAngⅡ+ACE2 group.
2.2 ACE2 siRNA干扰后各组VSMC AT1R的蛋白表达量ACE2的siRNA干预后,各组细胞培养3 d后,同样采用Western blot检测各组AT1R的蛋白表达量,结果显示:ACE2组AT1R的蛋白表达量较空白对照组、GFP组、脂质体组明显下降,差异具有统计学意义,提示ACE2能抑制AT1R的表达; si-ACE2 组和AngⅡ组的AT1R蛋白表达水平则明显高于空白对照组、GFP组和脂质体组;AngⅡ+si-ACE2 组的AT1R蛋白表达水平则高于AngⅡ组;AngⅡ+ACE2+si-ACE2组的AT1R蛋白表达水平则高于AngⅡ+ACE2组。以上各对比组差异表明随着ACE2蛋白表达的减少,AT1受体蛋白的表达能上调。
2.3 Western blot检测各组p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平从Fig 2A、2B可看出,各组细胞经过相关干预后培养72 h后,经Western blot检测p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平后发现: ACE2组p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平较空白对照组、GFP组及脂质体组明显低,差异具有统计学意义;AngⅡ+ACE2组的p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平则低于AngⅡ组,表明ACE2能抑制p-ERK1/2蛋白、p-STAT3蛋白磷酸化,而其表达的减少p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平能相应提高;si-ACE2组和AngⅡ组的p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平则明显高于空白对照组、GFP组和脂质体组;AngⅡ+si-ACE2组的p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平则高于AngⅡ组; AngⅡ+ACE2+si-ACE2组的p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平则高于AngⅡ+ACE2组,以上各si-RNA干扰组p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平较各对应无si-RNA干扰组明显升高,提示ACE2表达被抑制后能提升p-ERK1/2蛋白、p-STAT3蛋白磷酸化水平。
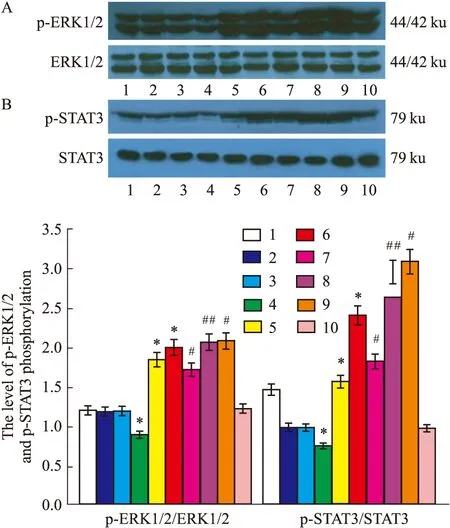
Fig 2 Level of p-ERK1/2 (A) and p-STAT3 (B) phosphorylation after ACE2 si-RNA interference in VSMCs measured by Western
1. Control group; 2. Lentiviral-GFP group (GFP); 3. Lipofectamine 2 000 group;4. Lentiviral-ACE2 group;5. Si-ACE2 group; 6. AngⅡ group;7. AngⅡ+lentiviral-ACE2 group; 8. AngⅡ+lentiviral-ACE2+si-ACE2 group; 9. AngⅡ+siACE-2 group; 10. Scrambled siRNA group.*P<0.05vscontrol,GFP and lipofectamine 2000 group;#P<0.05vsAngⅡ group;##P<0.05vsAngⅡ+ACE2 group.
3 讨论
肾素血管紧张素系统(RAS)主要存在于血液循环或组织中,在维持机体水、电解质平衡、调节血压及心血管系统细胞增殖过程中起重要的作用,其过度激活是许多心血管疾病始动因素。而AngⅡ作为RAS系统最重要的活性物质,通过与其特异性受体结合,可使血管平滑肌细胞增生、心肌细胞肥大,引起血管壁增厚、血管阻力增大、左心室肥厚、肾脏纤维化等病理改变。研究表明[5,6]AngⅡ主要与ATlR特异性结合,经ERK1/2、JNK/SAPK、p38MAPK、JAK2-STAT等信号路径调节血管内皮细胞的增殖、血管收缩以及氧化应激等病理生理反应[7]。目前研究表明ACE2主要通过两种路径拮抗ACE作用:(1)直接水解AngI产物AngⅡ成Ang(1-7),减少了AngⅡ浓度,进而减少AngⅡ对靶细胞刺激引起细胞增殖、肥大等病理生理改变; (2)AngⅡ降解产物Ang(1-7)具有舒张血管、降低血压、抑制血管平滑肌细胞增殖肥大、抑制心肌细胞的肥大、抑制心肌成纤维细胞的增殖等拮抗AngⅡ效应[8]。而Ang(1-7)结合其特异性受体Mas后通过蛋白激酶B(AKT)信号通路、MAPK通路、环单磷酸鸟苷/蛋白激酶G(cGMP/PKG)通路以及钙信号通路等实现上述拮抗AngⅡ的作用。Deng等[9]研究表明在自发性高血压大鼠的延髓头端腹外侧区中过表达ACE2明显降低了乙酰胆碱的释放,改善了血管内皮细胞的生理功能。本课题组前期研究中通过载有ACE2基因慢病毒表达载体转染大鼠平滑肌细胞,发现ACE2可以通过下调AT1R和/及ERK1/2的磷酸化水平而抑制平滑肌增殖,提示ACE2的过表达具有血管保护作用[10]。
然而,并不是ACE2表达越高就越好,在ACE2转基因动物模型研究表明,心肌过度表达ACE2后出现心脏传导阻滞及室性心律失常,可发展为室颤和猝死,且其严重程度与ACE2表达量成正比。Iwata等[11]研究表明,Ang(1-7)与心脏成纤维细胞上Mas受体结合,具有抗心肌肥大、抗纤维化及抑制胶原合成等作用,表明Ang(1-7)能逆转AngⅡ的不良作用,改善心肌重构功能;而在自发性高血压大鼠(SHR)、盐敏感高血压大鼠(SBH)、自发性高血压易卒中型(SHRSP)等高血压大鼠动物模型的QTL定位研究表明,ACE2基因为高血压相关基因,且在发生高血压后上述动物模型ACE2mRNA和蛋白均呈低水平表达;在肝星状细胞中研究表明,Ang(1-7)能够通过Mas受体抑制AngⅡ激活的ERK信号通路,达到抑制AngⅡ介导的肝脏纤维化[12]。此外多项研究表明,Ang(1-7)通过内皮细胞、产生NO、前列环素及激肽等血管活性物质起到扩张血管作用。以上研究表明ACE2蛋白表达的变化及其平衡在心血管疾病的发生、发展起着重要作用。
本研究利用RNAi技术,用si-ACE2干扰慢病毒转染ACE2基因的VSMCs后,运用Western blot检测各组AT1受体蛋白、ERK1/2蛋白、STAT3蛋白的磷酸化水平发现:si-ACE2干扰之后各组AT1受体蛋白的表达量、ERK1/2蛋白、STAT3蛋白的磷酸化水平较各对应无干扰组明显上升,而与之对应的ACE2蛋白的表达量相应减少。表明si-ACE2干扰之后随着ACE2蛋白表达的减少,其对AngⅡ诱导的AT1受体蛋白及其下游信号ERK1/2蛋白、STAT3蛋白的磷酸化抑制减弱。本研究结果与Gallagher等[13]研究结果相符,他们研究发现当ACE2表达下调之后AngⅡ浓度升高,通过AT1R诱导细胞ERK1/2蛋白、STAT3蛋白磷酸化水平的提高;此外亦有研究表明在肾小管上皮细胞NRK-52E中,Ang(1-7)通过Mas受体抑制高糖诱导的ERK1/2和p38磷酸化,进而抑制高糖刺激的上皮细胞向间充质细胞转化[14]。另外,本课题组在既往研究实验中用慢病毒转染SD大鼠血管平滑肌细胞,使ACE2基因过表达,发现其除能明显抑制AngⅡ诱导的VSMC增殖之外亦能抑制AT1受体蛋白表达以及AT1受体下游信号通路的STAT3蛋白、ERK1/2蛋白的磷酸化。上述研究表明,ACE2除了通过分解AngⅡ减少其浓度达到拮抗AngⅡ诱导血管平滑肌增殖作用,亦可以直接影响AT1受体蛋白表达及其下游信号MAPK磷酸化水平抑制AngⅡ作用。
本研究同时也发现,AngⅡ能抑制ACE2蛋白的表达,且AngⅡ与si-ACE2能协同抑制ACE2蛋白表达。Gallagher等[15]在大鼠小脑或延髓星形胶质细胞中研究中表明AngⅡ能够减少上述细胞中ACE2 mRNA和蛋白的表达50%以上,且该效应同样能被AT1受体阻断剂缬沙坦或者氯沙坦所阻断;而AT2受体阻断剂PD123319对ACE2 mRNA和蛋白的表达则没有影响。另外本课题组在早期研究中发现SHR大鼠用缬沙坦治疗后,SHR大鼠血压下降的同时,肾脏中ACE2蛋白表达亦明显上升并且呈剂量依赖性升高。此外,本研究发现AngⅡ组、AngⅡ+si-ACE2组ERK1/2蛋白、STAT3蛋白磷酸化水平高于各相应对比组。以上实验表明AngⅡ可能通过影响AT1受体及其下游通路ERK1/2蛋白、STAT3蛋白磷酸化水平调节ACE2蛋白的表达。
总之,本研究表明ACE2-Ang(1-7)-Mas与ACE-AngⅡ-AT1这两条轴之间在心血管系统相互调节、相互拮抗。除了通过ACE2分解AngⅡ减少其作用、生成Ang(1-7)与其特异性Mas受体结合拮抗AngⅡ引导的血管平滑肌细胞增殖、心肌重构等作用外,亦可以直接通过AT1R-ERK1/2或STAT3信号通路相互拮抗,共同发挥心血管系统的病理生理调节作用,对心血管疾病的治疗具有重要指导意义。
猜你喜欢
山东医药(2022年26期)2023-01-06
昆明医科大学学报(2022年3期)2022-04-19
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
黑龙江科学(2021年14期)2021-08-06
昆明医科大学学报(2021年3期)2021-07-22
中国CT和MRI杂志(2020年3期)2020-03-27
信息技术时代·上旬刊(2019年4期)2019-09-10
中国临床医学影像杂志(2019年4期)2019-06-18
分析化学(2017年12期)2017-12-25
