
α2肾上腺素受体与配体作用关键受体氨基酸位点研究进展
2020-03-16 05:31:48卢凤凤周培岚苏瑞斌
中国药理学与毒理学杂志 2020年11期
卢凤凤,周培岚,苏瑞斌
(军事科学院军事医学研究院毒物药物研究所,抗毒药物与毒理学国家重点实验室,北京 100850)
肾上腺素受体(adrenergic receptor,AR)包括α1-AR,α2-AR和β-AR,属于G蛋白偶联受体(G protein-coupled receptor,GPCR)超家族。α2-AR在药理学上又分为α2A,α2B和α2C3种亚型,分别位于10,2和4号染色体上。研究发现,啮齿类动物的α2D-AR在氨基酸序列上与人α2A-AR有高度同源性,是α2A-AR的种属变体,故被列为α2A-AR亚型[1]。
α2A-AR主要分布在中枢神经系统,如蓝斑外侧、臂旁核、脑桥核、海马旁回和大脑皮质等;α2B-AR主要分布在外周组织,如肾、肝、肺、心及血管组织等,在中枢神经系统中只存在于丘脑核团和脑干孤束核;α2C-AR主要分布在中枢神经系统,如纹状体、嗅球、海马和大脑皮质等,但表达量远小于α2A-AR[2-4]。研究表明,α2-AR介导的镇静、麻醉催眠等作用主要通过中枢α2A-AR发挥作用,但少量分布在脊髓的α2A-AR可介导镇痛;α2B-AR主要参与外周血管的收缩;α2C-AR参与调节多种脑功能,如感觉运动整合和应激反应[5]。
1 α2肾上腺素受体关键氨基酸位点
1.1 保守性关键受体氨基酸位点
1.1.1 3.32位天冬氨酸(aspartic acid,Asp)3.32关键位点
对于经典的保守性氨基酸残基,如α2-AR的Asp3.32在已知与胺配体结合的所有GPCR中是保守的,激动剂的阳离子氮在α2A-AR,α2B-AR和α2C-AR 3种亚型中与带负电荷的Asp羧酸盐侧链形成盐桥[6-7],儿茶酚胺类配体的β-OH基团与Asp残基之间相互作用,一系列咪唑啉衍生物配体的阳离子基团与Asp3.32相互作用[8]。若用天冬酰胺(asparagi⁃nate,Asn)取代Asp3.32则可消除受体以高亲和力结合育亨宾的能力,且降低激动剂抑制cAMP的作用。以上研究表明,α2-AR的Asp3.32在高亲和力配体结合及受体功能中至关重要。
1.1.2 丝氨酸(serine,Ser)5.42和Ser5.46关键位点
位于受体第5跨膜区(transmembrane 5,TM5)中的2个保守丝氨酸残基Ser5.42和Ser5.46与儿茶酚胺羟基形成氢键作用,Ser5.42与儿茶酚胺环的meta-OH基团相互作用,而Ser5.46通过para-OH基团形成氢键,有助于稳定活性受体的构象[7,9]。定点突变研究发现,α2-AR的Ser5.46与儿茶酚胺的对羟基结合,而β2-AR的Ser5.46与儿茶酚胺的间羟基形成氢键作用[10],但Ser5.46位点在 α1A-AR,α1B-AR,α1C-AR,α2A-AR,α2B-AR,α2C-AR,β1-AR,β2-AR和β3-AR 9种AR中高度保守[11-12]。Ser5.42和Ser5.46突变为半胱氨酸,增强α2-AR与UK14,304的部分亲和力,但降低儿茶酚胺激活α2-AR的能力[13]。
以上3个保守性氨基酸残基可形成受体的分子对接中心[6](图1),对于α2-AR 3种亚型受体的结构研究至关重要。
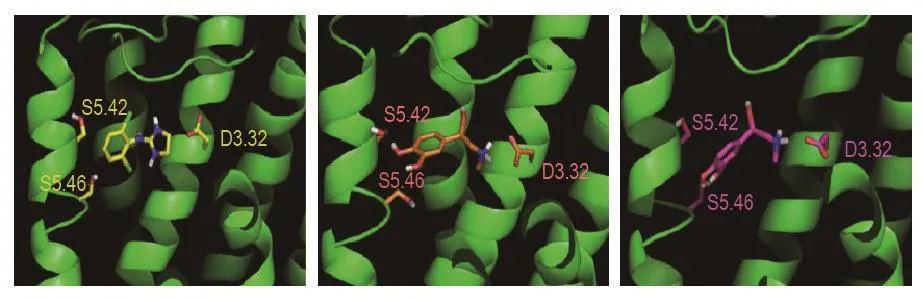
图1 α2A肾上腺素受体(左)、α2B肾上腺素受体(中)、α2C肾上腺素受体(右)与配体的分子对接图[6]
1.1.3 Asp2.50关键位点
Asp2.50在94%的A类GPCR中是保守的,且是跨膜区埋腔的一部分,尽管Asp2.50不直接参与与配体的结合,但已被证明参与信号转导及受体激活相关的构象变化[14-15],同时研究发现,Asp2.50附近的水分子保守簇与保守残基之间的相互作用参与受体激活[16-17]。Asp突变为Asn会降低α2-AR与激动剂的亲和力,且会导致受体与K+通道选择性解偶联[18]。以上提示Asp2.50参与α2-AR的功能变化,虽然目前该受体晶体结构尚未被解析,但Asp2.50可能是参与晶体结构解析并参与受体活化的微开关[19]。
1.2 非保守性关键受体氨基酸位点
近期研究表明,通过α2-AR 3种亚型受体的分子对接,发现某些非保守性氨基酸位点可能是α2-AR所特有的,并可能指导今后α2-AR的高选择性药物研发。
1.2.1 不同于 β肾上腺素受体的非保守性关键受体氨基酸位点
α2A-AR 的酪氨酸(tyrosine,Tyr)6.55、半胱氨酸(cysteine,Cys)5.43、苯丙氨酸(phenylalanine,Phe)7.39等非保守性氨基酸可能参与受体的功能发挥。Tyr6.55与肾上腺素、去甲肾上腺素等激动剂形成氢键,当Tyr6.55突变为Asn,取消了α2A-AR所介导的双重效应[20],而在β受体(β1和β2受体)中,该位置的残基是Asn,并认为Asn与其配体形成的氢键作用是激活β受体的标志[21]。UK14,304通过与暴露在结合腔中的Cys5.43的巯基侧链形成共价键,介导受体失活,而在Ser突变体中,受体与配体的亲和力降低[9]。α2A-AR的Phe7.39通过 π-π 键与胍基苯和UK14,304等激动剂相互作用[22-23],然而β2受体的Asn通过氢键作用与配体结合,当α2A-AR的Phe7.39突变为Asn时,对β2受体的2种选择性配体沙美特罗和沙丁胺醇无反应,提示Phe7.39位点有可能是α2-AR与激动剂结合的特异性位点[23]。
1.2.2 α2-AR 3种亚型间的非保守性关键受体氨基酸位点
位于第2个细胞外环(extracellular loop 2,ECL2)的谷氨酰胺(glutamine,Gln)以及Gln7.36等非保守性氨基酸可能为设计α2B-AR的选择性配体化合物提供新方向。α2A-AR和α2C-AR在ECL2的Asp通过带负电荷的侧链羧基与带正电荷的配体形成稳定的盐桥作用,而α2B-AR在ECL2的Gln可通过氢键与配体的羟基结合。位于α2B-AR的Gln7.36也存在相似的氨基酸关联,而位于α2A-AR和α2C-AR该位点的是带正电荷的赖氨酸,不能通过氢键作用与配体的羟基结合[23]。
α2C-AR选择性拮抗剂JP1302中带正电荷的胺基与AspECL2形成盐桥,并且与Tyr6.58形成阳离子-π相互作用,α2C-AR 具有由 AspECL2、精氨酸(argi⁃nine,Arg)ECL3和Tyr6.58形成的特殊交互网络,而该网络在α2B-AR和α2C-AR中不存在。α2C-AR三重突变体D206A/R409A/Y405T取消了选择性拮抗剂JP1302的拮抗功能,表明3个非保守性关键受体氨基酸位点对于α2C-AR的亚型选择性至关重要[11]。关于α2-AR亚型选择性的重要氨基酸位点仍需进一步探索。
2 基于 α2肾上腺素受体关键氨基酸位点的药物研究
2.1 基于关键受体氨基酸位点的药物筛选
研究发现,在α2A-AR突变体介导的钙离子反应中,激动剂右美托咪定和可乐定等对受体的最大激活幅度不受Asp2.50突变的影响,UK 14304和羟甲唑啉的最大幅度均显著降低,BHT 920可诱导更高的钙离子响应;拮抗剂在α2A-AR D79N突变体中对钙离子的响应值由强到弱顺序为:阿替哌唑>BRL 44408=咪唑克生≈SKF 86466>地塞米松。而在α2A-AR突变体T373K中,不同的激动剂与拮抗剂引起钙离子的反应与79位突变体有所不同[24],说明Asp79和373位苏氨酸在α2A-AR药物筛选中发挥关键作用,这为今后指导临床用药奠定理论基础。α2-AR除了介导镇痛、麻醉等药理作用外,本实验室研究还发现α2-AR也可介导致死性毒性作用(待发表),但目前针对该受体介导致死性毒性作用的关键氨基酸位点尚未报到,而针对该关键氨基酸位点的拮抗性药物筛选更是未见报道。
2.2 基于关键受体氨基酸位点的临床药物研究
对α2-AR选择性激动剂的镇痛和麻醉特性的研究已有几十年的历史,开发这些激动剂的临床应用仍然是一个令人感兴趣的领域,特别是作为疼痛治疗的佐剂和麻醉剂[25]。研究发现,α2-AR激动剂与阿片类激动剂在镇痛方面具有协同作用,这是临床疼痛管理中的一个重要特性,因为协同作用使剂量降低,可能使副作用最小化,尤其在治疗慢性阿片类不敏感疼痛状态方面[26-28]。与野生型小鼠相比,在D79N突变小鼠中,α2A-AR激动剂UK14303或右美托咪定与阿片类激动剂DAMGO联合应用,两者抗伤害或镇痛的协同作用消失,表明α2A-AR的Asp2.50对这种相互作用是必要的[29]。可乐定与右美托咪定的联用,在野生型小鼠中表现出显著的镇痛协同作用,然而在α2A-AR D79N突变小鼠中此协同作用消失,说明Asp79对于受体发挥镇痛作用至关重要,而且α2-AR激动剂除了与阿片受体激动剂联合应用外,不同的α2-AR激动剂之间也可联用[27]。目前,基于α2-AR关键受体氨基酸位点的临床药物研究较少,仍需进一步研究。
3 α2肾上腺素受体与配体相互作用的常用研究方法
3.1 受体与配体相互作用的结合口袋
α2-AR 3种亚型分别有由450,450和461个氨基酸残基组成,N端有2个糖基化位点,与一般GPCR结构相似,为7次跨膜结构,7条跨膜束由3条细胞内单环和3条细胞外单环首尾相连,7个疏水性跨膜区中的氨基酸序列在α2-AR的3种亚型受体中高度保守,但第3胞内环较长,其氨基酸序列在各亚型间存在较大的异质性,胞外区与配基识别有关[30]。每个跨膜区由20~26个氨基酸形成α螺旋,并形成对鉴定和结合配体分子至关重要的口袋。目前研究表明,疏水性氨基酸形成的“口袋”就是受体结合配体的关键部位,该“口袋”结构也被称为“结合口袋”。结合口袋是研究受体和配体相互作用的重点关注部位,决定着与配体结合的特异性。
3.2 同源模建和分子对接
获得该结合口袋处的对接信息可辅助确定与配体结合特异性有关的氨基酸位点。α2-AR作为GPCR超家族的成员之一,具有GPCR的一般特性,即不溶于水,自然丰度大,在溶剂中不易稳定存在,结晶难度较大[31]。晶体结构对明晰其与配体之间的相互作用意义重大,但α2-AR晶体结构尚未被成功解析。生物信息学在蛋白质空间结构预测及药物设计领域的应用有助于解决这一问题[32]。
具体研究方法有:①同源模建法:首先确定模板,采用单序列或多序列进行同源性比较,构建基于模板3D结构的靶标模型,获取建模蛋白序列的空间结构;② 分子动力学(molecular dynamics,MD)模拟:采用分子力学与MD的方法,根据物理化学基本原理,从理论上计算蛋白质的空间结构[33]。一般GPCR经过同源模建后需要进行MD模拟优化,MD模拟方法有2种,一种是先构建蛋白与脂水膜体系,然后用MD研究整个蛋白-膜-水体系;另一种是构建蛋白-水体系,主要用来研究配体结合性[34]。模型建立后,将目标受体与配体进行分子对接,是受体空腔与配体小分子进行相互识别的过程,它们的识别过程遵循几何能量匹配和化学环境互补的原则,首先通过搜寻受体分子结构的活性位点,将小分子置于活性口袋中,然后分子对接程序计算活性中心的各个方位,打分函数搜寻小分子的最佳结合方式[35]。生物信息学方法虽可高效率地预测目标蛋白与配体的相互作用模式,但确证这些关键受体氨基酸位点仍需定点突变这样的生物学方法进行验证。
4 展望
α2-AR是介导麻醉镇静、镇痛和催眠的重要靶点之一,明确其发挥生理及药理作用的关键受体氨基酸位点有助于α2-AR高选择性靶点药物研发,降低药物的不良反应,为今后在临床的药物应用奠定理论基础。α2-AR的保守性关键氨基酸位点研究逐渐偏向非保守性关键氨基酸位点,可能是未来研发亚型高选择性药物的微开关。目前,已有研究证明了α2-AR与其配体相互作用的某些关键受体氨基酸位点,但大多数研究主要关注单个氨基酸位点的关键性作用。实际上,受体结合口袋的不同氨基酸残基与不同配体之间的作用既有相似之处,又有差别,单一氨基酸的改变可能会影响受体构象的改变,也有可能需要多个氨基酸同时改变影响受体构象变化,从而引起受体功能的改变,联合突变研究可能为探索关键的受体氨基酸位点提供新反向。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
中成药(2018年10期)2018-10-26 03:41:22
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
医学研究杂志(2015年5期)2015-06-10 06:43:26
无机化学学报(2014年6期)2014-02-28 17:32:06
