
高血糖诱发卒中后出血转化的发生机制及治疗药物研究进展
2020-03-16 05:31:48刘楠楠魏广义王淑美孔令雷杜冠华
中国药理学与毒理学杂志 2020年11期
刘楠楠,魏广义,王淑美,孔令雷,杜冠华
(1.广东药科大学中药学院,广东 广州 510006;2.中国医学科学院北京协和医学院药物研究所,北京市药物靶点研究与新药筛选重点实验室,北京 100050)
卒中是一类严重危害人类健康的疾病,具有高发病率、高致死率、高致残率和高复发率的特点[1]。卒中分为出血性和缺血性卒中(ischemic stroke,IS),其中IS发生率约占70%。出血转化(hemor⁃rhagic transformation,HT)是指IS发生后脑内继发性出血的现象。HT的发生会增加IS的致死率和致残率,严重影响预后。临床研究表明,急性IS患者发病后24 h内HT发生率为15.19%,整个卒中期HT的发生率可达30.2%[2-3]。HT的发生过程可能是卒中的自然结果,也可能与抗凝药、溶栓药等药物的使用有关。除此之外,高血糖、高血压和高血脂等也是HT发生的重要危险因素。
高血糖可出现在卒中的不同病理阶段,发生率约为40%[4-5]。卒中后高血糖的原因比较复杂,除基础性疾病如糖尿病或代谢综合征外,患者发病前摄入高糖食物、发病后输注葡萄糖或使用皮质激素类药物也可能引起高血糖。此外,卒中过程中生理应激致使应激激素如糖源性儿茶酚胺、胰高血糖素和皮质醇等产生也可能引起高血糖。高血糖加重卒中后损伤,诱发HT发生,严重影响卒中的预后,但其诱发HT的机制尚不明确[6-7]。HT发生的原因较多,机制复杂。高血糖是卒中后HT发生的重要风险因素,其诱发的HT是临床常见的主要类型。此外,不同原因引起HT的机制不同,但其根本原因都是血脑屏障(blood-brain barrier,BBB)破坏和微血管损伤。因此,研究高血糖促进HT的发生机制,寻找新靶点,开发新的治疗药物和优化已有药物对于IS的治疗和预后具有重要意义。本文就近年来高血糖诱发HT的发生机制、潜在靶点及治疗药物的研究进行综述,以期为HT的临床预防和治疗提供参考。
1 出血转化的临床分类
临床上对HT的分类有不同方法,根据患者发生HT后是否有神经系统症状加重,可将HT分为无症状HT和有症状HT[8]。根据影像学变化,可分为出血性梗死(hemorrhagic infarction,HI)和脑实质血肿(parenchymal hematoma,PH)。具体又可分为HI-1型:沿梗死边缘的高密度小点状瘀点出血;HI-2型:梗死区域内融合性高密度的片状出血,无占位效应;PH-1型:有均匀性高密度血肿形成,占位效应轻,出血区域梗死面积≤30%;PH-2型:脑内血肿梗死面积<30%,且有明显占位效应。临床上,HI型的发生率高于PH型。
2 高血糖诱发出血转化发生的相关机制
高血糖诱发HT的发生是一个复杂的病理过程,涉及能量代谢障碍、氧化级联、炎症、血管异常和细胞凋亡等多种途径,作用于烟酰胺腺嘌呤二核苷酸/还原型烟酰胺腺嘌呤二核苷酸/去乙酰化酶1(nicotinamide adenine dinucleotide+/reduced nicotin⁃amide adenine dinucleotide/Sirtuin-1,NAD+/NADH/Sirt1)、低氧诱导因子1α/血管内皮生长因子/基质金属蛋白酶(hypoxia-inducible factor 1α/vascular endothelial growth factor/matrix metalloproteinases,HIF-1α/VEGF/MMP)、晚期糖基化终末产物/晚期糖基化终末产物受体(advanced glycation end products/the receptor of AGE,AGE/RAGE)、高迁移率族蛋白 1/Toll样受体 4/NF-κB(high mobility group box 1 protein/Toll-like receptor 4/NF-κB,HMGB1/TLR4/NF-κB)、凋亡诱导因子/胱天蛋白酶3(apoptosis-inducing factor/caspase 3,AIF/CASP-3)等通路和靶点,靶点之间也存在相互作用,直接或间接促进MMP表达,引起基底膜断裂,最终导致BBB和神经血管稳态的破坏,促进HT发生。本部分主要综述高血糖促进HT发生的病理机制,并在此基础上总结相关的通路和靶点(图1)。
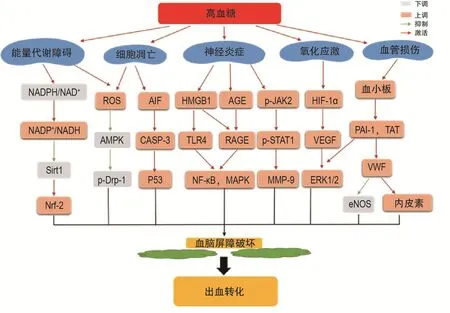
图1 高血糖诱发出血转化的靶点和通路.NADPH:还原型烟酰胺腺嘌呤二核苷酸磷酸;NAD+:烟酰胺腺嘌呤二核苷酸磷酸;Sirt1:去乙酰化酶1;Nrf-2:核因子相关因子2;ROS:活性氧;AMPK:腺苷酸活化蛋白激酶;p-Drp-1:磷酸化线粒体分裂蛋白1;HIF-1α:低氧诱导因子1α;VEGF:血管内皮生长因子;ERK1/2:细胞外调节蛋白激酶1/2;AGE:晚期糖基化终末产物;RAGE:晚期糖基化终末产物受体;HMGB1:高迁移率族蛋白1;TLR4:Toll样受体4;MAPK:丝裂原活化蛋白激酶;p-JAK2:磷酸化酪氨酸激酶2;p-STAT1:磷酸化信号传导子和转录激活子1;MMP-9:基质金属蛋白酶9;AIF:凋亡诱导因子;CASP-3:胱天蛋白酶3;PAI-1:纤溶酶原激活剂抑制剂1;TAT:凝血酶-抗凝血酶复合物;VWF:血管内皮生长因子;eNOS:内皮型一氧化氮合酶.
2.1 能量代谢障碍
大脑是对缺氧最敏感的器官,它的活动主要依靠葡萄糖有氧氧化提供能量,一旦缺血时间较长即可引起严重的不可逆损伤。因此,能量代谢障碍是卒中发生后的主要病理机制。脑组织在氧充足的情况下将葡萄糖代谢成二氧化碳和水,并产生ATP来维持大脑正常的生理活动,而卒中发生后,脑组织缺血缺氧,无氧糖酵解加强,导致大量乳酸堆积造成酸中毒,损伤神经元并诱导自由基在脑内过度聚集。高血糖促进无氧糖酵解过程,导致细胞内醛糖堆积,脑血管内皮细胞缺氧加重,进而使细胞内蛋白被不可逆糖基化并引起细胞内电位变化,刺激活性氧(reactive oxygen species,ROS)过度生成,诱发氧化级联反应,引起MMP等蛋白异常表达,破坏BBB。
能量代谢障碍导致多个通路和靶点的功能异常。还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotin⁃amide adenine dinucleotide phosphate,NADPH)是一种辅酶,通常作为生物合成的还原剂,不能直接进入呼吸链氧化。NADPH通过将H+转移到烟酰胺腺嘌呤二核苷酸NAD+,然后以NADH的形式进入呼吸链,自身被氧化为NADP+。细胞内葡萄糖水平的增加导致NADPH过度氧化为NADP+,NAD+加速还原为NADH,引起ROS过度产生,刺激脑内氧化级联反应,导致脑内NAD+缺乏和Sirt1含量降低。Sirt1是一个高度保守的蛋白去乙酰化酶家族蛋白,广泛参与能量代谢、应激反应和细胞衰老/凋亡等过程[9]。研究表明,高糖血症卒中大鼠和糖尿病卒中大鼠体内Sirt1表达均明显下降,同时Nrf-2表达下降,NF-κB乙酰化增加,MMP-9表达增加,高血糖可能通过抑制Sirt1表达,加重氧化应激(oxi⁃dative stress,OS)和神经炎症,损伤 BBB,诱发HT[10-11]。综上所述,高血糖通过促进缺氧后的糖酵解过程,导致能量代谢相关靶点功能异常,诱导神经炎症的发生,加重IS损伤,促进HT的发生。
2.2 血管损伤
血管内皮细胞是BBB的重要组成部分,血管内皮层的完整性直接影响BBB的正常功能。高血糖引起卒中后梗死周围血管改变,大脑皮质血管间的弯曲、密度和络脉增加,血管壁增厚,同时刺激血管内皮细胞释放多种凝血因子,导致血液处于高凝状态[12-13]。高血糖促进细胞内糖酵解,激活蛋白激酶C通路,引起舒血管物质和缩血管物质失衡,导致毛细血管内皮肿胀和血管管腔直径变小。同时缺血半暗带区血流量减少,红细胞瘀滞,最终引起血管破裂,HT发生[14]。
血小板在机体凝血过程中发挥重要作用,活化的血小板既可直接参与血栓形成,也可通过刺激内皮细胞上的P选择素表达,诱导炎症反应,进而促进凝血因子如I型纤溶酶原激活剂抑制剂(plasmino⁃gen activator inhibitor-1,PAI-1)、凝血酶-抗凝血酶复合物等的表达,并抑制组织纤溶酶原激活剂的活化,阻止机体溶栓,形成血栓。研究表明,PAI-1升高可作为预测HT发生的风险因子。高血糖可直接引起牛主动脉内皮细胞PAI-1的表达,抑制NF-κB可减少PAI-1的表达,表明高血糖可通过NF-κB通路调节PAI,参与体内凝血过程[15-16]。血管内皮生长因子(vascular endothelial growth factor,VEGF)作为一种多效性生长因子,与血管生成、神经再生、轴突可塑性、神经元存活和血管通透性有关,在IS发生前期,VEGF可增加内皮的通透性,使内皮细胞与周围平滑肌细胞紧密性变差,BBB完整性被破坏。此外,MMP-2和MMP-9表达的增加会导致基底膜断裂和紧密连接(tight junction,TJ)蛋白降解,破坏BBB完整性[17]。VEGF可调节MMP的表达和活性,二者均与IS急性期的BBB渗透性增加密切相关。综上所述,高血糖可以通过促进IS发生前期的血小板活化和PAI-1表达,刺激血栓更快形成;同时促进VEGF和MMP表达,引起血管损伤,导致BBB的破坏。
低氧诱导因子(hypoxia-inducible factor,HIF-1)是一种具有转录活性的核蛋白,其靶基因涉及低氧适应和炎症等。VEGF和MMP作为HIF-1下游基因,与血管通透性和细胞外基质的降解密切相关。脑缺血时,细胞缺氧直接刺激HIF-1α的表达,同时细胞无氧糖酵解的加速导致细胞酸中毒和ATP酶活性丧失,间接上调HIF-1α水平,并促进其与下游基因的相互作用,导致血管内皮受损,通透性增加,BBB损伤。高血糖促进IS大鼠脑微血管中HIF-1α和VEGF表达,与正常血糖IS大鼠相比,高血糖组BBB损伤更加严重[18]。通过特异性敲除内皮细胞HIF-1α基因或用胰岛素控制血糖,可改善糖尿病大鼠的BBB渗漏和脑梗。此外,细胞外调节蛋白激酶1/2与VEGF高表达后的血管生成障碍和血管炎症密切相关,可能导致血管受损[19]。因此,高血糖对血管的直接损伤作用、促血栓形成和促炎作用共同导致了血管的损伤及BBB结构和通透性的改变,促进HT的发生。
2.3 炎症损伤
卒中后伴随复杂的神经炎症反应,包括脑内胶质细胞的激活、外周炎症细胞的浸润和炎症因子的释放,涉及NF-κB和丝裂原活化蛋白激酶(mito⁃gen-activated protein kinase,MAPK)等炎症通路的调节[20]。研究表明,高血糖促进脑缺血后内皮黏附分子细胞间黏附分子1和血管细胞黏附分子1的表达,引起中性粒细胞浸润到脑实质,加重神经炎症[21]。过氧化物酶体增殖活化受体γ(peroxisome proliferator-activated receptor gamma,PPARγ)是核激素受体家族中的配体激活受体,控制许多细胞内的代谢过程。糖尿病小鼠IS后,PPARγ的活性降低,导致紧密连接蛋白降解。高血糖促进IS小鼠单核/巨噬细胞的促炎性极化,而激活PPARγ可抑制炎性极化,证明PPARγ与高血糖诱导的炎症相关[22-23]。此外,高血糖引起IS大鼠脑组织内非受体酪氨酸激酶2(Janus kinase 2,JAK2)和信号转导与转录激活因子(signal transducer and activator of transcription 1,STAT1)蛋白磷酸化,促进肿瘤坏死因子α,白细胞介素1β和MMP-9的表达,提示高血糖可能通过JAK2/STAT1途径损伤脑组织导致BBB损伤和HT发生[24]。
AGE及其受体RAGE以及HMGB1在高血糖诱导的炎症损伤中发挥重要作用[25]。研究发现,AGE和RAGE的结合可能是引起糖尿病血管病变和血管功能障碍的关键因素。IS发生后,高血糖导致糖酵解加速,细胞内蛋白质被不可逆糖基化为AGE,促进AGE与RAGE结合,导致神经炎症和血管内皮损伤[26]。HMGB1是一种高度保守的非组蛋白核DNA结合蛋白,由坏死细胞被动释放或由巨噬细胞、髓样树突状细胞和自然杀伤细胞主动分泌。IS发生后,高血糖刺激HMGB1释放,与其受体RAGE和Toll样受体2、4结合,进而促进NF-κB入核和MAPK信号通路蛋白磷酸化,诱发神经炎症[27]。此外,HMGB1和AGE表达升高刺激白细胞介素6、早期生长应答因子1和激活蛋白1等多种炎症细胞因子释放,释放的炎症因子刺激平滑肌细胞增生和迁移,分泌大量细胞外基质,诱导粥样斑块形成,刺激凝血因子和组织因子释放,加速血栓形成和血管破坏[26-27]。高血糖通过多种机制加重IS后神经炎症,导致血管内皮受损,加重BBB损伤,引起HT。
2.4 细胞凋亡
线粒体参与细胞代谢、生长、分化和稳态,维持正常细胞功能,线粒体损伤促进ROS生成,导致细胞凋亡、细胞周期异常和复制延迟。细胞凋亡涉及到B细胞淋巴瘤2家族中抗凋亡和促凋亡蛋白之间的平衡以及线粒体通透性转换孔(mitochondrial permeability transition pore,MPTP)的调节。高血糖加重IS引起的线粒体损伤,包括减少细胞增殖和ATP生成及改变线粒体膜电位等,诱导线粒体碎片化、空泡化和嵴破坏,导致神经元产生过多的ROS,引起内质网应激并诱导MPTP持续不可逆的开放,最终驱动细胞色素c(Cytochrome-c,Cyt-c)等线粒体内容物释放到细胞质中[28]。
凋亡诱导因子(apoptosis-inducing factor,AIF)是一类存在于线粒体内外膜间隙的黄素蛋白,在正常生理状态下,作为线粒体氧化还原酶催化Cyt-c和NAD+之间的电子传递,当细胞受到凋亡刺激时,引起细胞染色体凝聚及DNA片段化。研究表明,高糖暴露显著增加内皮细胞AIF和Cyt-c向细胞质的释放,激活P53信号通路,引发内皮细胞胱天蛋白酶依赖性凋亡;同时,大量氧自由基的产生破坏细胞膜中的蛋白质和脂质,也会导致细胞凋亡的发生[29]。
高血糖打破IS后线粒体诱导的细胞凋亡与线粒体自噬的平衡,加重脑卒中损伤。动力蛋白相关蛋白1(dynamin-related protein 1,Drp-1)在生理条件下主要存在于细胞质中,而在应激条件下广泛表达并转位到线粒体,激活线粒体分裂和凋亡。线粒体融合素2(mitofusin-2,MFN2)基因调节线粒体自噬。AMPK是一种对能量波动敏感的丝氨酸-苏氨酸激酶,调节一系列信号转导过程,在能量稳态中发挥重要作用,调节细胞凋亡和线粒体自噬,可通过磷酸化Drp-1来抑制细胞凋亡。高血糖抑制IS诱导的神经元损伤后Drp-1磷酸化以及MFN2和AMPK的表达,促进细胞凋亡,打破凋亡与自噬之间的平衡,导致BBB损伤加重。同时,AMPK和ROS的生成密切相关,提示高血糖损伤BBB和促进HT发生,可能与ROS/AMPK/Drp-1通路异常相关[30]。线粒体损伤导致细胞内环境的改变,引起细胞凋亡和自噬的发生,加重脑损伤,促进HT的发生。
2.5 其他
在高血糖促进HT发生的病理过程中还涉及其他机制,包括兴奋性毒性、细胞内钙超载和脑血流灌注减少等。高血糖加重能量代谢障碍,导致缺血区兴奋性神经递质释放增加,激活谷氨酸受体,导致钙内流和钙依赖通路异常激活,最终造成细胞内钙超载。高血糖导致的脑血管功能异常也使IS后脑血流减少,颅内压升高,脑血管压力增大,更易破裂和出血。
高血糖促进HT的发生涉及上述多种机制和靶点,这些机制之间相互影响,作用于不同靶点。其中血管功能异常是高血糖加重BBB损伤的重要因素,高血糖导致的血管形态异常、血小板活化、血栓形成和OS间接损伤血管内皮功能都可能导致血管异常和BBB结构破坏。高血糖通过多种机制促进IS后神经炎症的发生,包括促进中性粒细胞浸润和小胶质细胞极化,激活HMGB1和NF-κB等炎症通路,释放多种炎症细胞因子直接引发神经炎症,通过影响能量代谢产物以及凝血因子等表达间接促进炎症的发生。此外,高血糖引起的线粒体功能障碍是导致能量代谢异常和细胞凋亡发生的重要原因。能量代谢异常使乳酸等代谢产物堆积和ROS过度生成,造成细胞酸中毒、OS和神经炎症的发生。因此,高血糖导致的血管异常、神经炎症和线粒体功能障碍共同作用,导致BBB结构破坏,诱导HT发生。这些病理机制涉及的靶点众多,有些靶点已在动物实验中证实与HT的发生相关,但目前尚无进一步的临床试验证据。同时,由于HT的病理机制复杂,同时干预多个靶点可能产生更好的治疗作用,这也是未来药物研发的重要方向。
3 高血糖诱发出血转化的治疗药物
IS过程中HT发生涉及血栓形成、血管堵塞破裂和红细胞外流至脑组织,临床治疗主要通过手术机械取栓或t-PA溶栓。IS发生后立即取栓或溶栓,可减缓血管压力,预防血管压力过大导致HT的发生。高血糖诱发HT的发生机制与血糖水平较高导致的一系列反应有关,针对高血糖促进HT发生的病理机制而采取的治疗策略,如抗炎、抗氧化和抑制氧化级联反应等可发挥一定的治疗作用。在防治高血糖诱导HT的药物研究方面,已有研究报道的一些能抑制高血糖引起的HT的潜在药物见表1。
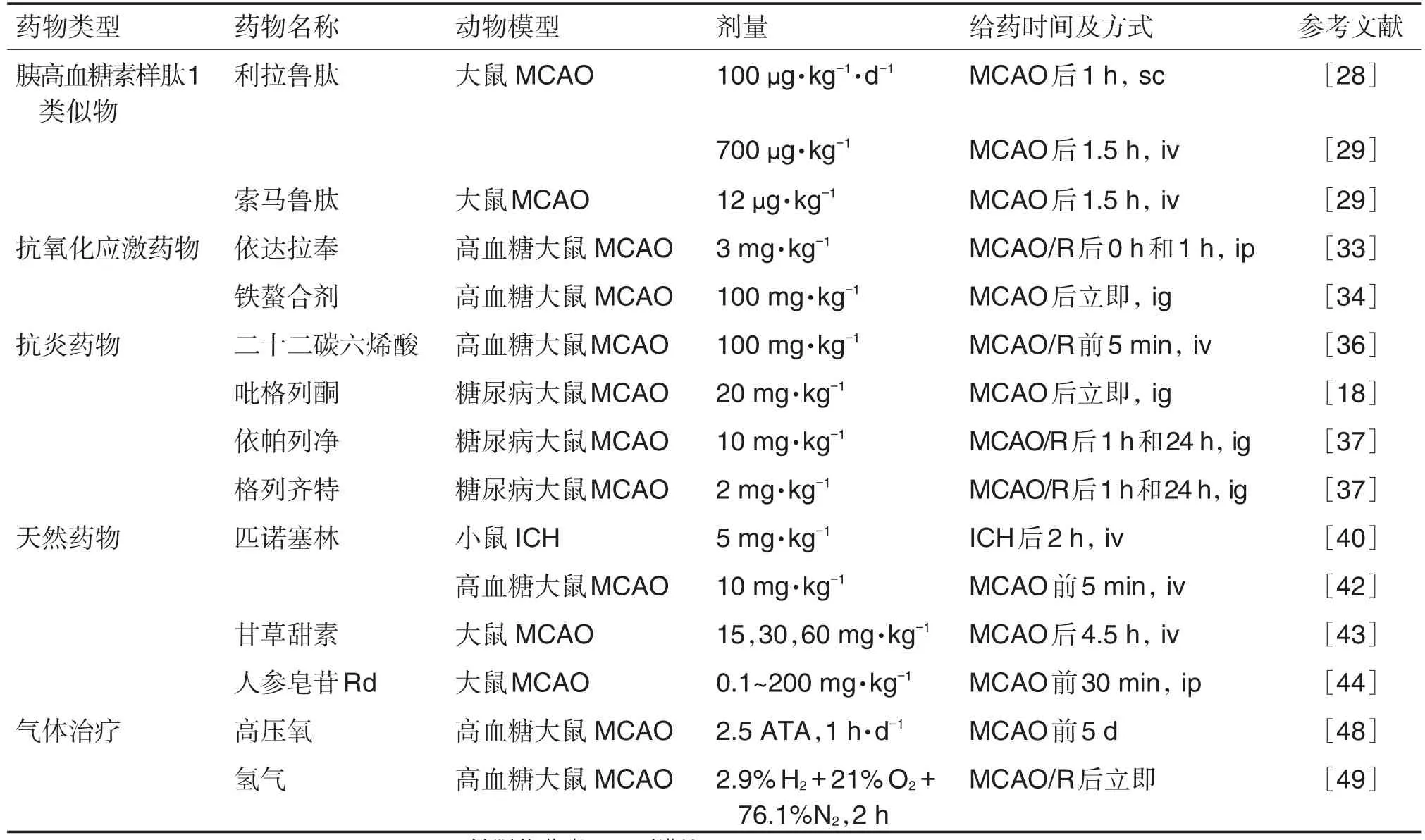
表1 出血转化治疗药物
3.1 控血糖药物
卒中患者在急性IS后,表现出较高的胰岛素抵抗和血糖水平,这与卒中后较差的临床疗效和预后密切相关。因此,在IS治疗中,考虑能否采用控制血糖的治疗方法以及通过控制血糖有效改善预后是临床预防和治疗HT的重要策略。
控血糖药物主要有胰岛素、胰高血糖素样肽1(glucagon-like peptide-1,GLP-1)及其类似物等。胰岛素可间接抑制血小板聚集、凝血和血栓形成,抑制促炎因子表达。卒中后立即进行胰岛素治疗能缓解与高血糖相关的血管反应,如血管收缩舒张失衡和血管内皮迅速破坏等。此外,胰岛素还可通过增加神经元和星形胶质细胞中一氧化氮合酶(nitric oxide synthase,NOS)的表达和NO的生成,引起血管舒张,改善血流[31]。利拉鲁肽(liraglu⁃tide)是一种长效GLP-1类似物,动物实验发现,在大鼠MCAO后1 h,通过sc给予利拉鲁肽100 μg·kg-1可以减少脑梗死,其机制可能是通过减少ROS,激活磷脂酰肌醇激酶/蛋白激酶B和MAPK通路对缺血脑组织发挥神经保护作用,减少HT的发生[32]。大鼠MCAO后1.5 h iv给予利拉鲁肽350,700和1050 μg·kg-1也可保护脑组织,降低HT发生率[33]。索马鲁肽(semaglutide)12 μg·kg-1也具有相同的作用。因此,GLP-1类似物对于急性IS患者,特别是正在接受溶栓治疗的患者,能有效预防出血。
但近年来的多项研究表明,卒中后控制血糖可增加低血糖概率。由于卒中后依靠糖酵解供能的特点,低血糖并不利于卒中治疗,甚至增加患者死亡率。因此,临床实践中应考虑适度降低血糖水平并密切监测以避免低血糖发生。同时建议结合患者血糖水平,当血糖水平>8.6 mmol·L-1时进行血糖控制治疗,并在治疗手段上建议使用iv给予胰岛素,而不是迅速降低血糖水平,避免产生过山车效应[34-35]。基于上述研究,降糖药物在卒中治疗中的应用还有待进一步的探讨和考察。
3.2 抗氧化应激药物
高血糖再灌注过程中,能量代谢障碍导致NADPH氧化酶和内皮型NOS增加。二者的增加促进自由基生成,激活MMP-9,诱导基底膜降解,破坏BBB的完整性,加重脑损伤和HT的发生。因此,清除自由基,抑制氧化应激反应也是HT治疗的有效手段。
依达拉奉(edaravone)是一种强效自由基清除剂,多项研究表明,其对IS脑损伤有一定的缓解作用[36]。在高血糖卒中动物模型中发现,大鼠ip给予依达拉奉3 mg·kg-1,可通过清除自由基维持血管基底膜的结构,抑制出血性梗死的发生[37]。铁螯合剂去铁铵(deferoxamine,DFX)具有神经保护作用,大鼠ig给予DFX 100 mg·kg-1能直接清除羟基自由基并抑制细胞周期转变,减少脑梗死[38]。另外,DFX能通过HIF-1调节神经保护基因,如HO-1和红细胞生成素等,保护BBB。右美托咪定(dexmedeto⁃midine,Dex)是一种高选择性α2-肾上腺素受体激动剂,可降低交感神经活动,具有镇静和镇痛的作用,临床上作为麻醉辅助药物使用。研究发现,在链脲佐菌素(streptozocin,STZ)诱导的糖尿病小鼠脑缺血后,立即ip给予Dex 25 μg·kg-1可通过抑制氧化应激、炎症和细胞凋亡来减缓脑损伤[10]。在小鼠脑出血(intracranial hemorrhage,ICH)模型上,Dex能通过抑制出血后氧化应激反应保护脑组织,可能与抑制PPARγ共激活因子1α失活有关[39]。目前,抗氧化应激药物对于缓解高血糖IS损伤已有较多实验研究,且发现多种有效药物,提示抗氧化应激药物可作为后续研究的重要方向。
3.3 抗炎药物
神经炎症贯穿于高血糖卒中的发生发展过程,OS会刺激多种炎症细胞因子的释放,血小板活化诱发单核巨噬细胞促炎性极化,凝血因子PAI-1的产生可介导中性粒细胞和内皮细胞的相互作用,这些因素共同导致了炎症反应的发生。炎症反应可直接或间接刺激MMP的释放,破坏基底膜,造成BBB损伤,因此抗炎是HT治疗的主要措施。
神经保护素D1的前体二十二碳六烯酸(doco⁃sahexaenoic acid,DHA)是一种主要的Omega-3脂肪酸,具有辅助脑细胞发育、抗衰老和改善血液循环等作用。研究发现,在IS中高血糖模型大鼠再灌注前静脉注射DHA 10 mg·kg-1可降低HT,减少梗死体积,改善神经功能,还可抑制ICAM-1介导的炎症反应,抑制Ⅳ型胶原降解,稳定BBB[40]。吡格列酮(pioglitazone)既具有降糖作用,又能激活单核巨噬细胞的内源性调节剂PPARγ,抑制单核巨噬细胞向非炎性极化。口服吡格列酮20 mg·kg-1可显著增加STZ诱导的高血糖脑出血大鼠动物模型中PPARγ的表达,减少炎性单核巨噬细胞,抑制神经炎症,缓解脑出血[22]。依帕列净(empagliflozin)是一种新型的抗糖尿病药物,格列齐特(gliclazide)是一种磺酰脲类口服抗糖尿病药[41]。口服依帕列净10 mg·kg-1和格列齐特2 mg·kg-1可显著减轻STZ模型大鼠的脑缺血损伤,两者可选择性地抑制近端肾小管中钠-葡萄糖协同转运蛋白2,从而增加尿中葡萄糖的排泄,降低血糖,减少大脑OS,抑制与肥胖相关的神经炎症,保护脑组织。抗炎药物的研究已取得了一些进展,但它们的特异性并不好,寻找特异的抗高血糖IS后神经炎症的药物仍是值得认真研究的问题。
3.4 天然药物
高血糖诱导HT的发生涉及多种机制和多个靶点,靶点之间独立或协同发挥作用,寻找能同时作用于多个靶点,调控多种机制的药物应作为HT药物研发的重点。天然药物作用特点为多靶点多功效,且具有一定的安全性和应用基础,是治疗高血糖诱导HT药物的重要来源。
匹诺塞林(pinocembrin)是一种天然的黄酮类化合物,存在于蜂蜜、蜂胶、姜根和野生马郁兰等植物中,具有神经保护作用[42]。研究发现,在t-PA诱导的HT模型中,匹诺塞林可显著降低脑内MMP-2和MMP-9的表达,促进紧密连接蛋白的表达,保护BBB[43]。在ICH模型中,匹诺塞林可通过NF-κB途径抑制神经炎症反应并改善脑出血现象[44]。此外,匹诺塞林还可通过调控自噬减缓IS损伤[45]。近期研究也发现,匹诺塞林10 mg·kg-1能减轻高血糖诱导的HT,降低出血的发生率和死亡率[46]。甘草甜素(glycyrrhizin)是中药甘草中的一个成分,具有抗炎、抗氧化和抗兴奋毒性作用。在t-PA诱导的大鼠HT模型中,甘草甜素可通过抑制过氧亚硝酸盐和超氧化物的形成,介导HMGB1/TLR2信号通路,调节一氧化氮合酶和NADPH氧化酶亚基的表达,抑制MMP-9,保护BBB,减少HT的发生。因此,甘草甜素可能对高血糖诱导的HT也有潜在的治疗作用[47]。人参皂苷Rd可抑制2,3-DHBA和2,5-DHBA的羟基自由基形成以及诱导型NOS的过度表达,显著改善脑缺血大鼠神经功能[48]。此外,人参皂苷Rg1对脑缺血再灌注损伤也具有一定的保护作用,可通过抗氧化和抑制细胞凋亡保护脑组织[49],具有潜在的抗高血糖诱导HT的作用。以上研究结果表明,天然产物通过多途径作用于高血糖诱导的HT病理过程,发挥抗HT的作用,可以作为未来HT防治药物的研究重点。
3.5 气体
近年来,医用气体作为神经保护剂的应用已引起医学界的广泛关注。医用气体包括氧气、氢气、硫化氢和NO等[50]。研究发现,在创伤性脑损伤、卒中、蛛网膜下腔出血和神经退行性疾病等不同类型脑损伤动物模型中,医用气体均有一定的神经保护作用,其中研究较多的有高压氧和氢气。
高压氧治疗是在超过一个大气压的环境中呼吸纯氧治疗脑外伤的一种辅助治疗方法,可通过多种途径保护脑组织[51]。高血糖大鼠在脑缺血前每天给予1 h的2.5 ATA高压氧,持续5 d,可通过调节NLRP3炎症小体的表达[41],改善卒中后的炎症反应,也可通过激活环氧合酶2释放15脱氧前列素J2,促进PPARγ和视黄酮X受体α形成异二聚体,减缓炎症反应,抑制MMP-9的过度激活,保护BBB[9]。高压氧还可通过ATP/NAD+/Sirt1通路介导恢复早期能量供应,减少细胞死亡,也能通过抑制水通道蛋白4过表达,减少脑水肿,保护BBB[52]。氢通过抑制脑组织内小胶质细胞的促炎性极化发挥脑保护作用。进一步研究发现,H2保护BBB的机制可能是H+与氯、氟和羟基自由基等氧化元素发生反应,在缺血性脑损伤后起到抗氧化作用[53]。另外,H2还可调节MMP的表达,保护内皮细胞和BBB。但目前气体治疗在卒中的应用研究并不深入,可能仅发挥辅助作用。气体治疗与药物联用可能具有协同作用,值得进一步研究。
4 结语
卒中前高糖食物的摄入、患者既有的代谢性疾病和卒中后的应激反应等都有可能导致高血糖。卒中过程中高血糖可能是一种或多种因素引起,可导致卒中后HT的发生,从而影响卒中的预后。高血糖诱导HT的发生机制复杂,各种机制之间独立或相互作用加重脑损伤。线粒体功能障碍同时会导致细胞凋亡和能量代谢障碍以及ROS的过度生成,进而促进超氧化物生成,刺激HMGB1释放,促进炎症反应。PAI-1既参与血管障碍机制,又通过促进NF-κB入核加重炎症反应。在高血糖损伤机制中,多种损伤会进一步引发炎症反应,使脑组织处于炎性环境中。因此,抗炎是HT防治的一个重要研究方向。
可根据高血糖诱发HT发生的机制发现不同的作用靶点,寻找相应的防治药物。调节血糖类药物虽能改善血糖水平,但其使用条件受限,且对HT的作用尚需进一步明确。抗氧化和抗炎药物在动物实验中取得了一定的治疗效果,但仍需临床试验验证。天然药物具有多靶点和作用广泛的特点,使之成为抗HT药物开发的热点,而气体治疗联合药物治疗也是未来HT防治探索的重要方向。
此外,抗HT药物的动物评价模型限制了药物的开发。动物模型的缺点在于人与动物的生理差异,导致某些药物在动物体内的有效性在临床试验中得不到重复,甚至无效。因此在未来研究中,寻找更加接近临床病理特点的动物模型和治疗方案极为重要。临床中,HT的诱发原因复杂,且致死率高,使得对HT的研究困难重重,这也是HT研究迫在眉睫的主要原因。随着研究的不断深入,HT的发生机制和作用靶点会越来越清晰,临床上HT的防治方案也会越来越成熟。
猜你喜欢
中老年保健(2021年3期)2021-12-03 02:32:25
海洋通报(2021年1期)2021-07-23 01:55:14
自我保健(2021年4期)2021-06-16 07:36:46
生物学通报(2021年4期)2021-03-16 05:41:26
中国生殖健康(2020年7期)2020-12-10 07:48:51
中国卫生标准管理(2015年15期)2016-01-15 02:58:45
中国医学装备(2015年10期)2015-12-29 12:00:30
医学研究杂志(2015年7期)2015-06-22 11:01:36
中国当代医药(2015年23期)2015-03-01 02:05:41
同位素(2014年2期)2014-04-16 04:57:16
