
阿尔茨海默病动物模型建立及评价研究分析
2020-03-16 05:31谢丹妮李劲松
中国药理学与毒理学杂志 2020年11期
邓 婷,谢丹妮,王 平,李劲松,徐 颖,孙 涛
(成都中医药大学1.中药材标准化教育部重点实验室,2.西南特色中药资源国家重点实验室,四川 成都 611137;3.广元市第一人民医院,四川 广元 628017)
阿尔茨海默病(Alzheimer disease,AD)是一种多发于老年群体的神经退行性疾病,最初由德国医生Alois ALZHEIMER于1907年发现并命名[1]。据统计,全球约有>5000万AD患者,预计到21世纪中期将增加到≥1.52亿,全球医疗总费用达到8180亿美元,对医疗体系、社会经济造成沉重负担[2]。AD主要临床表现为早期记忆力逐渐下降、轻微认知损伤,中后期记忆力明显下降,并出现语言和运动障碍及人格改变等。AD主要标志性病理学特征包括神经元胞外β淀粉样蛋白(β-amyloid,Aβ)聚集成老年斑(senile plaques,SP)、tau蛋白异常磷酸化形成神经原纤维缠结(neurofibrillary tangles,NFT)和神经元丢失等。由此,根据其病理特征提出相关发病机制的假说,包括氧化应激(oxidative stress,OS)损伤、tau蛋白异常磷酸化、胆碱能损伤、铝中毒和Aβ毒性等,并在相关假说的基础上建立AD动物模型,但目前尚缺乏一种能全面模拟AD病症表现的模型。
基于此,本文总结了OS损伤、胆碱能损伤、铝中毒、Aβ毒性和转基因5类AD动物模型的发病机制、建模方法和指标评价,并进一步总结各模型的特点,从而为构建符合临床特征且覆盖AD病症表现的AD动物模型提供思路。
1 氧化应激损伤模型
研究显示,自由基诱导的OS损伤在AD的发病机制中占有重要作用,自由基与其清除剂之间的不平衡,即氧化剂水平增加或抗氧化剂水平降低会引发OS[3]。自由基是在其外轨道上具有单个不成对电子的原子或分子。自由基中不成对的电子使其非常不稳定,能与蛋白质、DNA和脂肪酸等其他分子发生反应,导致细胞损伤、功能障碍,最终导致细胞死亡[3]。另一方面,自由基的产生也与Aβ[4]和晚期糖基化终末产物(advanced glycation end prod⁃ucts,AGE)[5]有关。同时,OS也可促进Aβ的产生并使其在脑内聚积,进而导致神经元退行性病变[4]。其动物模型主要有D-半乳糖(D-galactose,D-gal)模型和快速老化小鼠模型。
1.1 D-半乳糖模型
研究表明,长期大量皮下或腹腔注射D-gal后可转化为半乳糖醇,并在细胞内积累,引起活性氧(reactive oxygen species,ROS)增多,超氧化物歧化酶(superoxide dismutase,SOD)活性下降,导致细胞OS损伤;此外,D-gal也会形成AGE,促进肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、白细胞介素(interleukin,IL)-6和IL-1释放导致炎症反应,从而影响学习和记忆功能[6]。此模型出现与AD临床表现相同的症状,如认知功能减退和学习记忆下降,但未发现SP和NFT等病理学特征。同时,造模过程中存在较多不确定因素,如给药方法、周期、剂量等因素。此外,另有研究采用D-gal联合亚硝酸盐造模,促进了细胞的OS损伤[7]。具体造模方法及评价指标见表1。
1.2 快速老化小鼠模型
20世纪80年代,TAKEDA等[8]培育出一种快速老化小鼠(senescence accelerated mouse,SAM),包括快速老化亚系P系(SAMP)和正常衰老R系(SAMR)。多数P系表现出与年龄相关的退化,且研究证明SAMP8和SAMP10小鼠具有寿命较短、学习记忆缺陷、情绪障碍和昼夜节律改变的表征[9];此外,SAMR系SAMR1小鼠具有正常老化的特征,如生理指标及平均寿命均与正常衰老小鼠相似,因此常作为SAMP系小鼠的对照[10]。已有大量研究表明,与SAMR1小鼠相比,SAMP小鼠的各种组织(包括脑)的氧化状态更高[11],如丙二醛(malondial⁃dehyde,MDA)含量增多、SOD活性降低、谷胱甘肽(glutathione,GSH)含量减少等指标变化。同时,SAMP系小鼠也表现Aβ沉积、tau蛋白过度磷酸化、胆碱功能障碍和神经元丢失等AD病理特征[11]。SAM小鼠在正常发育过程中表现出较早的发病和不可逆的衰老进程,其脑组织形态结构的病变与AD的临床特征相似[12]。
2 胆碱能损伤模型
中枢胆碱能损伤学说认为,胆碱功能与记忆认知功能相关。有学者发现,AD患者大脑皮质和海马区的胆碱乙酰转移酶(choline acetyltransfer⁃ase,ChAT)和乙酰胆碱酯酶(acetyl-cholinester⁃ase enzyme,AChE)活性升高,导致乙酰胆碱(ace⁃tylcholine,ACh)合成减少[13]。同时,研究证明AD发病与胆碱能受体密切有关,如胆碱能终末的烟碱型受体和毒蕈碱型受体数量减少会降低认知功能[14-15]。另外,脑组织内缺乏神经生长因子(nerve growth factor,NGF)、脑源性神经营养因子(brain derived neurotrophic factor,BDNF)均能引起胆碱能神经元合成ACh的功能失常,进而导致记忆功能下降[16]。
采用不同方法损害大脑胆碱能系统来制备AD模型,可导致记忆减退和认知障碍,如东莨菪碱诱导模型。该模型通过向小鼠ip给予东莨菪碱阻断M胆碱受体,从而阻断中枢胆碱能神经功能使ACh减少[13],动物出现记忆力下降、认知功能减退等改变。此模型操作简易,但缺乏典型病理改变,如Aβ沉积和tau蛋白磷酸化等[13]。另外,也常结合胆碱能损伤和OS损伤这2种致病机制复合造模,有利于较全面地模拟AD临床症状,如东莨菪碱联合D-gal[15]。具体建模方法见表 1。
3 铝中毒模型
铝是一种大量存在的神经毒素,其易在AD大脑皮质和海马等区域积聚,长期接触铝会导致动物的氧化应激损伤和记忆障碍[17-18]。研究揭示,铝也能诱导炎症反应、引起轴突运输和突触结构异常,导致记忆严重丧失[18]。此外,铝参与β淀粉样前体蛋白(amyloid-β protein precursor,APP)的代谢途径,利于形成Aβ寡聚体及炎症斑块[17]。
该模型的建立是基于“铝中毒假说”。因此,相关研究通常采用大鼠口服三氯化铝(AlCl3)的方法造模,表现出铝含量、AChE活性、APP蛋白表达和炎症因子水平明显提高[17-18]。但此模型的诱导周期较长,且给药方法、剂量等不确定因素较多。另外,研究结合D-gal[19]和东莨菪碱[20]等多种物质复合造模,诱导动物出现OS损伤、胆碱能功能受损等多种发病机制,具体方法见表1。
4 A β毒性模型
Aβ是由APP代谢产生,APP经β-和γ-分泌酶连续剪切后形成长度在39~43个氨基酸的短肽。其长度在羧基端末端变化,主要产生Aβ40和Aβ422种分子,Aβ42的疏水性和聚集性强于Aβ40,因此毒性更大。1984年,首次在AD患者脑内发现Aβ氨基酸顺序与SP中的淀粉样物质为同系物[21]。因此,学者认为AD脑内SP的形成与Aβ有关。Aβ毒性假说认为,Aβ的积聚导致自由基损伤、tau蛋白磷酸化、递质丢失和炎症反应等,最终引起神经元功能失调和死亡、斑块形成和NFT等病理变化。
目前,研究人员通过向动物单侧或双侧脑室内分别注射Aβ1-42,Aβ1-40或Aβ25-353种淀粉样蛋白的寡聚体建立模型,注射部位有海马[23,28]、侧脑室[24]和基底前脑[25]等,其神经毒性与Aβ积聚相关,导致炎症因子IL-1β和TNF-α分泌增多[22,24,28],氧化水平提高及神经递质合成减少[23,26]。另外,利用Aβ联合D-gal[24,27-28]和鹅膏蕈氨酸[29]复合造模,较全面模拟AD的临床症状。具体建模方法见表2。经脑室内注射Aβ制备AD模型在一定程度上验证了Aβ毒性假说。不同研究对注射Aβ后导致的行为学和神经病理学检测指标不一致,这些差别可能与注射部位、剂量、状态、时间及留针时间等因素有关。侧脑室注射Aβ是一种常见的模拟AD的动物模型,可诱发与AD类似的症状,如学习记忆下降和炎症反应等。同时,也具有手术操作简单、创伤性小、定位准确、造模周期较短、成功率高等特点,但注射Aβ快速造模的方法与AD脑内Aβ缓慢沉积的病理变化不相符。
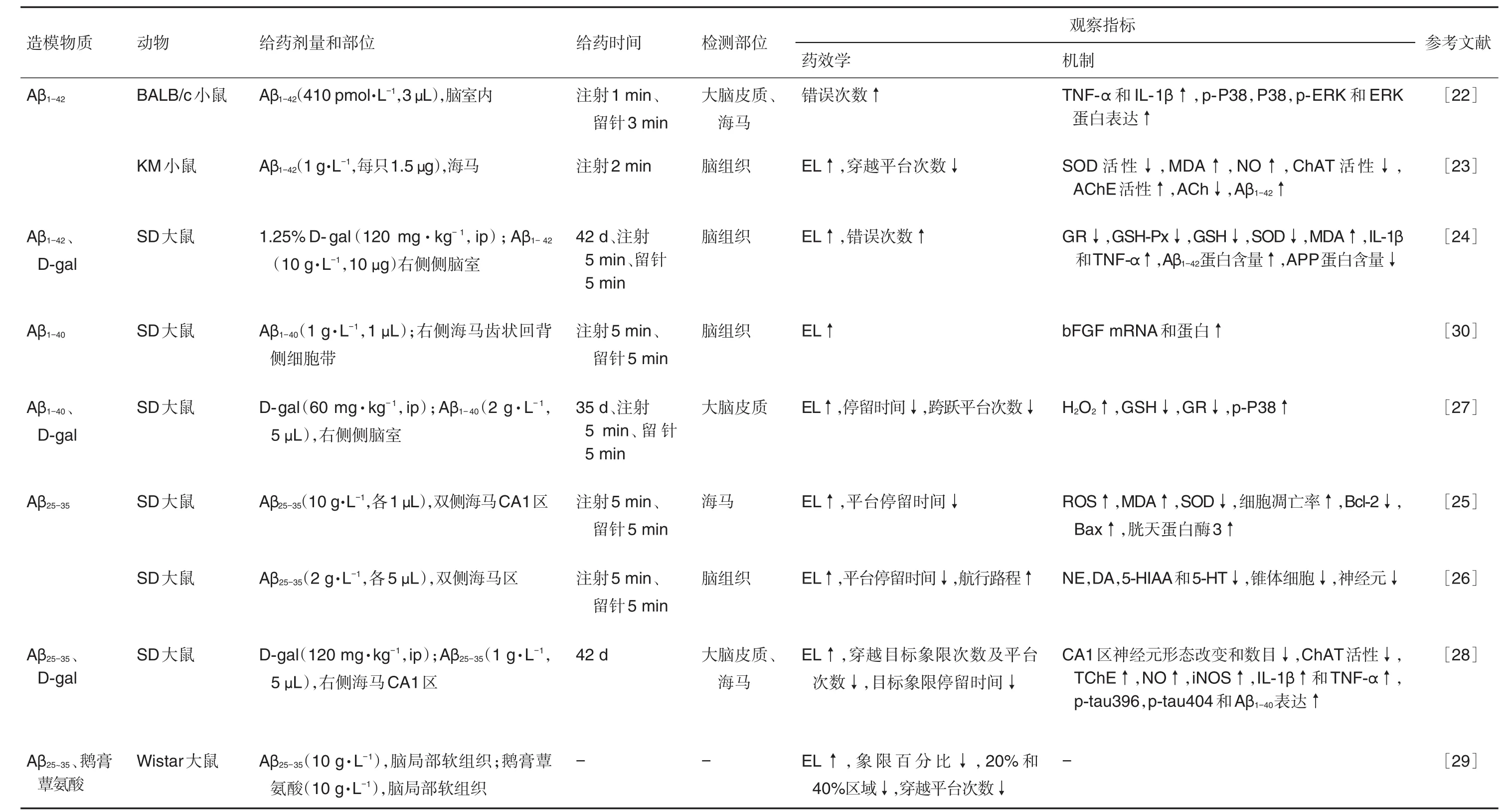
表2 β样淀粉蛋白(Aβ )毒性模型的建立和评价指标
5 转基因动物模型
转基因动物是研究AD发病机制和建立模型的一种较理想的工具,AD的相关基因包括APP基因、早老素(presenilin,PS)-1基因、PS-2基因、载脂蛋白E(apolipoprotein E,ApoE)基因及tau基因。其中APP、PS-1和PS-2基因与Aβ的生成有关,当APP基因发生突变时,APP不能被α-分泌酶正确识别并切割,使其更易被β-分泌酶和γ-分泌酶分解产生Aβ[31]。而PS基因被敲除后将影响神经发育及功能维持,导致神经祖细胞数量减少[32];同时,PS是γ-分泌酶的核心成分,该基因突变也会影响γ-分泌酶活性,导致Aβ42/Aβ40比值增大[33]。ApoE基因最先因参与脂质代谢而受到关注,随后被发现也参与AD。ApoE含有ApoE2,ApoE3和ApoE43个等位基因,其中ApoE4是晚期AD的主要遗传危险因素,可与Aβ结合形成不溶的高分子复合物,导致体外形成淀粉蛋白单纤维[34]。tau基因突变可导致tau蛋白表达水平异常和结构紊乱,从而出现tau蛋白异常磷酸化及神经缠结等AD病理特征。另有文献报道,AD也与人朊蛋白基因有关,朊蛋白是Aβ寡聚体的高亲和力受体[35],因而该基因的突变将导致Aβ寡聚体增多。根据上述突变基因已成功建立许多转基因模型,本文总结了部分模型的转基因构建体及相关病理特征,具体见表3。
至今,学者利用不断完善的转基因技术建立多种AD转基因模型,模型由单转基因向双转基因甚至三转基因发展,如表达APP基因的PDAPP单转基因小鼠[36]出现淀粉样沉积物;而APPSWE/PS1dE9双转基因小鼠[37]同时表达APP基因的瑞典型突变(K670N/M671L)和PS-1(PS1dE9)基因,出现斑块沉积和神经元丢失;三转基因模型(3xTg-AD)是可同时在AD相关脑区出现斑块和神经缠结病理的转基因模型[38]。因此,多转基因动物模型的特点在于可把多个相关致病基因结合在一起,较全面模拟AD患者脑内Aβ沉积、斑块形成和神经元丢失等病理特征。另外,多转基因动物也可较早出现行为异常,如双转基因5XFAD小鼠在1.5月龄时Aβ42开始沉积,同时伴有神经胶质增生[39];3xTg-AD小鼠6月龄时出现胞外Aβ沉积物,并在12个月时很明显[38];9月龄时出现tau病理性磷酸化,并在12个月逐渐增加[40]。
随着转基因技术日趋成熟,学者借助转基因技术将与AD相关的人类突变基因转入动物中,通过改变动物遗传学性状达到建立AD动物模型的目的。它能部分模拟人类AD患者Aβ沉积、神经炎症反应、神经缠结以及神经元丢失等部分神经病理特征,并表现出与AD临床相似的行为学障碍。
6 结语与展望
尽管AD研究进程不断深入,目前仍缺乏能准确表征及全面覆盖的模拟AD主要病理、生化及行为学等全部特征的理想动物模型,与临床AD患者的上述病症特点之间还存在一定差距,具有局限性,因此亟需开发更切合临床患者自身生物学特性的AD动物模型。当前应用于构建AD动物模型的方法较为多样(表1~表3),且各具特点,研究者可根据不同实验目的及损伤机制选取代表性动物模型。其中转基因动物模型是目前研究AD较为理想且应用广泛的模型,能表征多个病理特点或较早出现的病理改变,表现与AD患者类似的临床症状,因此转基因动物模型具有非常广阔的研究及应用前景。此外,AD动物模型的评价方法大多采用行为学指标,如逃避潜伏期和穿越平台次数等;组织病理切片;生化检测指标,如Aβ沉积和tau蛋白磷酸化(包括APP,Aβ和PP2A等指标)、胆碱能损伤(包括ChAT,AChE和ACh检测)、OS损伤(ROS,GSH,GSH-Px,MDA和其他氧化指标检测)、炎症反应(包括炎症因子TNF-α,IL-1β,IL-2,IL-4和IL-6等检测)和神经元丢失(神经元数量检测)。同时,查阅大量的相关文献,笔者发现较少研究采用病理检测指标,导致AD病变区域的关键病理特征检测缺失。
综上所述,选择适合的动物模型对探知AD的发病机制及病理特征具有重要价值,但仍有部分问题亟待解决。如缺乏与临床微观病理特点完全契合的动物模型建立方法,且已有建模方法尚不能达到高度的可重复性;缺乏统一的模型标准评价体系,反映实验动物学习记忆能力受损的神经生理和
运动行为的评价指标单一,相关理化指标检测尚不全面、不统一。因此,笔者认为在进一步的研究中应优化AD动物模型的建模及评价方法,并参考AD临床诊断标准对动物模型的相关指标进行评价,还可借助正电子发射型计算机断层显像、磁共振成像等先进影像学诊断技术综合构建AD动物模型多维度标准化评价方法,以期为AD动物模型的标准化评价提供思路与借鉴。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
中南药学(2022年4期)2022-07-04
世界科学技术-中医药现代化(2021年5期)2021-11-05
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2020年2期)2020-07-25
天津中医药(2019年9期)2019-09-18
中国疼痛医学杂志(2018年7期)2018-01-14
中外玩具制造(2013年8期)2013-11-25
