
N-甲基哌嗪烷基双吲哚马来酰亚胺蛋白激酶C抑制剂的合成研究
2020-03-15 16:24魏鹏东黎金海
世界最新医学信息文摘 2020年6期
魏鹏东,黎金海
(广州医科大学附属第三医院,广东 广州)
0 引言
随着生命科学研究的飞速发展,恶性肿瘤细胞内的信号转导、细胞周期的调控、细胞凋亡的诱导、血管生成及细胞与胞外基质的相互作用等各种基本过程正在被逐步阐明[1]。蛋白激酶C (Protein kinase C, PKC)是由Nishizaka等人于1977年首次发现的一组磷脂依赖性的蛋白丝氨酸/苏氨酸激酶,是细胞信号转导中重要的酶,广泛存在于动物和微生物等有机体内。一系列研究表明,许多疾病的发生都与PKC的异常表达有关,已经有大量关于从植物、微生物以及动物的次生代谢产物中发现PKC抑制剂的报道[2-4]合成和发现新的PKC抑制剂,特别是对亚型选择性好的PKC抑制剂具有重要的意义。我们设计合成的N-甲基哌嗪双吲哚马来酰亚胺蛋白激酶抑制剂可以为以后合成相关的PKC抑制剂及活性的研究打下基础。
1 仪器及试剂
1.1 实验仪器
旋转真空蒸发器、薄层色谱板、恒温磁力搅拌器、低温磁力搅拌器、回流冷凝管、烧杯、分液漏斗、烧瓶、试管、铁架台、MAT95XP 型高分辨率质谱仪、VarianMercury-Plus300型核磁共振仪等等。
1.2 试剂
3-溴-1-丙醇、对甲基苯磺酸吡啶盐(PPTS)、二氯甲烷、吲哚-3-乙醛酸甲酯、四氢呋喃、丙酮、N-甲基哌嗪、甲醇、乙醇、浓盐酸、乙酸乙酯、叔丁醇钾、氯化钠、吲哚-3-乙酰胺等。
2 实验步骤
2.1 3-溴-1丙醇四氢吡喃醚的合成
室温下将3-溴-1丙醇(5g,40mmol)与对甲基苯磺酸吡啶盐(Ppts)(1g, 4mmol)加入到干燥的二氯甲烷(50mL)中,在快速搅拌下于30min内滴入二氢吡喃(5.04g,60mmol),滴加完毕后室温继续反应,待原料充分反应完全后,加水充分搅拌,分离出有机相并用二氯甲烷萃取水相两次,合并有机相,用无水硫酸钠干燥,旋转真空蒸发器去除溶剂后进行真空蒸馏。
反应路线

2.2 N-羟丙基四氢吡喃醚-吲哚-3-乙醛酸甲酯(C)的合成
室温下将化合物N-吲哚-3-乙醛酸甲酯(2.0g, 9.85mmol)加入到经4A分子筛干燥过的DMF中(30mL),充分搅拌5min后加入碳酸铯(3.0g, 9.85mmol),室温下继续搅拌10min后将反应体系置于冰浴中搅拌5min,待体系温度降至0-5℃后,缓慢滴加3-溴-1-丙醇四氢呋喃醚(A)(2.0g, 9.85mmol)。滴加完毕,升至室温,原料药反应完全后停止反应。
产品用柱色谱来进一步提纯。筛选合适的洗脱液,以石油醚:乙酸乙酯=1:1作洗脱液洗脱。过柱一直使用TLC监测,当发现有明显点时,改用小试管收集,对各试管分别进行点样,确定其中所含有的物质,含相同物质的可以合并,最终得到红褐色产物2.1g,收率为69%。
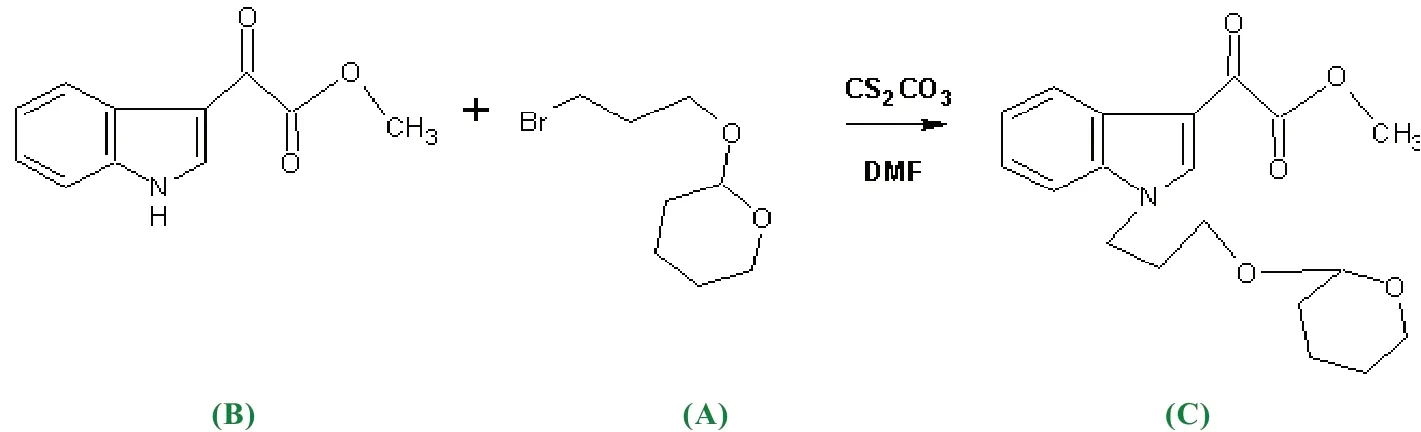
2.3 3-[1-(3-羟基丙基)-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的合成
将 吲 哚-3-乙 酰 胺(0.50g,2.87mmol)和 化 合 物(C)(1.19g,3.45mmol)加入到四氢呋喃(10mL)中,然后于0 ℃时慢慢加入叔丁醇钾(8.61mL,8.61mmol),加完后室温继续反应,TLC监测,反应约3h,该产物为(D)。冷却条件下再加入浓盐酸去掉保护基团(37%,10mL),继续反应约1h。将反应液倒入水中,用稀盐酸调pH至弱酸性,乙酸乙酯提取。合并有机相,水洗涤至中性,饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤[5,6]。
产物(E)用柱色谱纯化,以二氯甲烷:无水甲醇=10:1的洗脱剂经行洗脱得到红褐色产物0.82g,产率为62.1%。反应路线

2.4 3-[1-(甲磺酸基丙基)-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的合成
将化合物(E)加入到约5mL二氯甲烷中(二氯甲烷已经用分子筛干燥过了,可加入少量四氢呋喃,加速样品溶解),再加入吡啶及甲磺酸酐(CH3SO2)2O,在室温下搅拌下反应,TLC监测,约3h反应完全。最后反应得到化合物(F)。粗产物用柱色谱纯化,以二氯甲烷:无水甲醇=20:1为洗脱剂经行洗脱得到红褐色产物(F)0.62g,收率为71%[5]。
反应路线

下图是3-[1-(甲磺酸基丙基)-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的MS图谱:准分子离子峰486.1与M+Na(486.49)相符。

2.5 3-[1-[3-(4-甲基哌嗪)丙基]-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的合成
向 F (0.12g,0.25mmol)的DMF(5mL)溶液中加入 N-甲基哌嗪(0.65mL,6.25mmol),在65℃条件下反应24h,分别加入乙酸乙酯(25mL),水(10mL),分出有机层,再经饱和食盐水洗(10mL×2),无水硫酸镁干燥,过滤,旋干,得红色油状液,经硅胶柱层析(V甲醇:V二氯甲烷=1:9~1:4),得红色粉末固体71.2mg,收率61%。1H NMR (DMSO-d6, 300 MHz) δ: 1.76 ~1.91 (m, 2H), 2.10 (t, J=6.49 Hz , 2H), 2.16 (s, 3H), 2.21~ 2.43 (m, 8H), 4.23 (t, J= 4.74 Hz, 2H),6.56 (d, J=7.77 Hz , 1H), 6.60~6.75 (m, 2H), 6.84 (d, J=7.84 Hz,1H), 6.88~7.05 (m, 2 H), 7.34 (d, J=6.84 Hz, 1H), 7.45 (d, J=7.94 Hz, 1H), 7.69 (s, 1H), 7.76 (s, 1H), 10.89 (s, 1H), 11.66 (bs, 1H);MS m/z: 468 [M+H]+ 。
3 结果
N-甲基哌嗪烷基双吲哚马来酰亚胺化合物是吲哚马来酰亚胺类化合物的衍生物之一,我们以吲哚3-乙醛酸甲酯作为原料,经5步反应合成了N-甲基哌嗪烷基双吲哚马来酰亚胺,其结构经由MS和1HNMR图谱得到确证。
反应路线


N-甲基哌嗪烷基双吲哚马来酰亚胺蛋白激酶C抑制剂氢谱
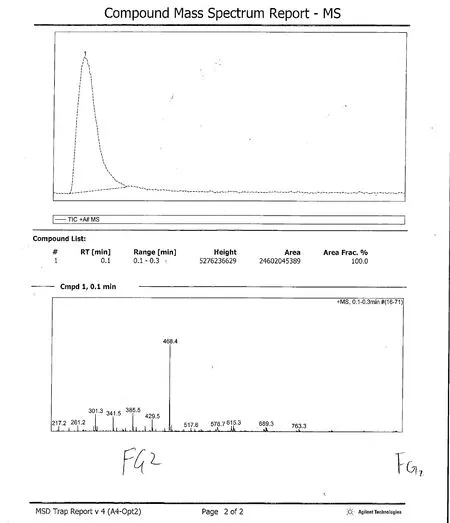
N-甲基哌嗪烷基双吲哚马来酰亚胺蛋白激酶C抑制剂质谱(MS)
准分子离子峰468.4,与M+H的分子量468.6相符[5-8]。
4 讨论
4.1 3-溴-1丙醇四氢吡喃醚的合成
第一步为成醚反应,PPTS为催化剂,用四氢吡喃醚保护醇羟基,以免影响到后续反应。
4.2 N-羟丙基四氢吡喃醚-吲哚-3-乙醛酸甲酯(D)的合成
第二步为亲核取代,DMF必须用分子筛干燥,保证在无水条件下,防止化合物乙醛甲酯基团水解;体系需在低温下进行,以免原料和产物发生分解;每隔1-2小时,TLC点板,找出最佳的时间反应。反应中加入碳酸铯作为催化剂,使吲哚N成为负离子发生亲核取代反应[9]。
4.3 3- [1-(3-羟基丙基)-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的合成
第三步为偶合成环反应,要在强碱叔丁醇钾催化作用下进行。加入叔丁醇钾后颜色变红,而且逐渐加深,加完叔丁醇钾后,反应液颜色已经变成紫红色。后面加入浓盐酸,反应液颜色没有太明显的变化。点板监测反应进程,因为考虑到脱保护后有—OH,所以产物极性偏大,TLC跑板跑到比较慢。
4.4 3- [1-(甲磺酸基丙基)-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的合成
第四步为醇甲磺酸化反应,加入二氯甲烷已经用分子筛干燥过了,可加入少量四氢呋喃,加速样品溶解。但是,由于原料中存有马来酰亚胺的胺基,容易与甲磺酸酐(CH3SO2)2O反应,故一定要控制好温度和配料比。这步实验为较严格的操作,各反应物和溶剂均不可含水,因为水与甲磺酸酐发生剧烈反应。为尽量避免甲磺酸酐在称量过程中与空气中的水分反应,可用估量法加入一半量。即用小容量瓶称取实放量的两倍,再估量地快速加入一半。
4.5 3-[1-[3-(4-甲基哌嗪)丙基]-3-吲哚]-4-(3-吲哚)-1-H-吡咯-2,5-二酮的合成
第五步反应为亲核取代反应,甲磺酸基是较好地离去基团,被N-甲基哌嗪取代得到目标产物。反应时间较长,需要TLC监控反应情况。
4.6 目标产物的合成意义
本研究合成的N-甲基哌嗪烷基双吲哚马来酰亚胺是一种新的PKC抑制剂,实验经改进和探索,我们的合成路线产率较高,合成时间较短,合成条件较成熟。该化合物经光环化反应还可以转变成吲哚咔唑类化合物,吲哚咔唑类化合物既是PKC抑制剂,又是细胞周期检测点抑制剂,有望开发成抗肿瘤药物。
5 结论
本文主要内容是利用药物设计的基本原理对双吲哚马来酰亚胺类化合物进行设计、合成。作者以吲哚-3-乙醛酸甲酯作为起始原料,首先与四氢吡喃保护的3-溴丙醇进行N-烷基化反应,然后与吲哚-3-乙酰胺偶合得到双吲哚马来酰亚胺化合物,接着进行甲磺酸化反应,最后与N-甲基哌嗪进行亲核取代反应得到目标化合物N-甲基哌嗪烷基双吲哚马来酰亚胺。目标化合物的结构经MS、1HNMR确认。另外,我们对N-甲基哌嗪双吲哚马来酰亚胺化合物合成路线进行了优化和改进,例如反应的最佳温度,配料比等反应条件对反应的影响,以提高反应的收率。目标化合物从结构上来看它应该有较好的亲水性,这些性质对于药物来说非常重要。目标化合物对热不稳定,应该在低温条件下保存。
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
林业工程学报(2022年1期)2022-02-26
中国医院用药评价与分析(2022年1期)2022-02-21
椰城(2021年9期)2021-09-13
看世界·学术下半月(2020年5期)2020-09-10
当代化工(2020年8期)2020-09-09
民用飞机设计与研究(2020年1期)2020-05-21
健康必读·下旬刊(2019年5期)2019-06-03
数学大王·中高年级(2016年10期)2016-09-10
分析化学(2015年7期)2015-07-30
