
SVIP 通过调节自噬抑制CCl4诱导的肝纤维化的研究
2020-03-14 02:55余光银陶丽丽殷晓敏陈耀丽王柳
中国现代药物应用 2020年3期
余光银 陶丽丽 殷晓敏 陈耀丽 王柳
肝纤维化是一种常见的病理状态,其中肝星状细胞(HSC)被激活,然后细胞外基质(ECM)蛋白积聚,通常与由感染、药物、代谢紊乱或自身免疫失衡引起的慢性肝病相关。肝纤维化如果控制不好,将导致不可逆的肝硬化,甚至是肝细胞癌[1,2]。到目前为止,除了去除潜在的病因或肝移植外,还没有有效的临床疗法来抑制肝纤维化的病理进展。除此之外,研究人员过分关注抑制肝星状细胞的活化,而不是保护肝脏的功能。毕竟,持续的肝实质细胞死亡对于引发瘢痕形成至关重要[3,4]。因此,需要研究肝纤维化的分子基础,并开发一种保护实质细胞和逆转肝纤维化的新治疗方法。自噬是一种关键的细胞内途径,破坏的细胞器和受损蛋白质被降解,为真核细胞中的细胞稳态提供能量。自噬途径通过保守的基因产物的几个阶段进行。在诱导(营养物剥夺或饥饿)后,雷帕霉素靶蛋白复合物1(mTORC1)的抑制激活ULK1/ 2-Atg13-Atg101-FIP200 复合物(Atgs,自噬相关基因),其启动分离膜或形成噬菌体。自噬的启动还伴随着Vps34-Vps15(p150)-Beclin1 复合物的激活。刺激Beclin1 复合物产生磷脂酰肌醇-3-磷酸(PI3P),其促进自噬体膜成核。自噬体延伸需要Atg5-Atg12 和微管相关蛋白1 轻链3/酵母自噬相关基因8(LC3/ Atg8)缀合系统[5,6]。此外,LC3 参与选择性转运自噬底物蛋白62(p62)/ SQSTM1 和NBR1 等蛋白质,这些蛋白质含有特殊的LC3 相互作用区域基序,用作细胞结构隔离的适配器,如线粒体,蛋白质聚集体和其他细胞结构。GTP 酶Ras 相关蛋白7(Rab7)需要完成与溶酶体融合的自噬体阶段。在最后阶段,溶酶体酸水解酶降解自噬体内容物,并释放自溶酶体的内容物用于代谢回收。自噬在调节脂肪形成中起关键作用,与脂肪变性和肝纤维化有关。它可以通过脂质减少脂滴。否则,长期脂质负荷可能在体外和体内改变膜脂组成并减少自噬体和溶酶体的融合[7]。因此,在肝脏中过量脂质抑制自噬可能导致肝细胞中脂滴积聚。然而,据报道,活化的自噬通过降解脂滴为肝星状细胞的活化和增殖提供能量。自噬通过降解胶原蛋白来抑制纤维化。自噬的激活降解小鼠肝脏中的Ⅰ型胶原蛋白,减少氧化应激,并抑制炎症以抑制纤维化。它还可以保护肝细胞免受凋亡。因此,自噬在肝星状细胞活化和从脂肪变性到纤维化的过程中的作用是有争议的。
1 资料与方法
1.1 方法 在饥饿模型中,在对照组(禁食)中,对15 只7 周龄小鼠进行4 h 禁食,1 h 再喂养的模式,之后立即处死。在实验组(肝纤维化)中,对15 只小鼠注射1 ml/kg 的50%CCl4橄榄油2 次/周,持续8 周,对照组注射相同剂量的橄榄油8 周。对于肝纤维化和禁食模型中,注射CCl4或橄榄油,将处理过的大鼠禁食1 h。之后收集肝组织和血清进行分析。在饥饿期间,这些动物可以获得饮用水。使用三唑试剂从细胞颗粒或新鲜组织中提取总核糖核酸。如前所述,通过MTT 分析评估细胞活力。经过不同的处理(DMSO 或0.05%CCl4)和不同的培养时间(0、24、48、72 h),在测试波长和参考波长为570、630 nm 的扫描孔微培养板读取器中读取孔的吸光度。HepG2细胞在4℃下用10%甲醛固定30 min,用BODIPY493/503 染色15 min。用磷酸缓冲液(PBS)洗涤后,细胞核用DAPI 染色20 min。荧光显微镜检测脂滴和自噬体,进行图像采集和图像分析。
1.2 统计学方法 采用GraphPad Prism 第6 版统计学软件处理数据。数据表示为至少三个独立实验的平均值标准划分,两组以上数据比较进行单向方差和事后多重分析。P<0.05 表示差异有统计学意义。
2 结果
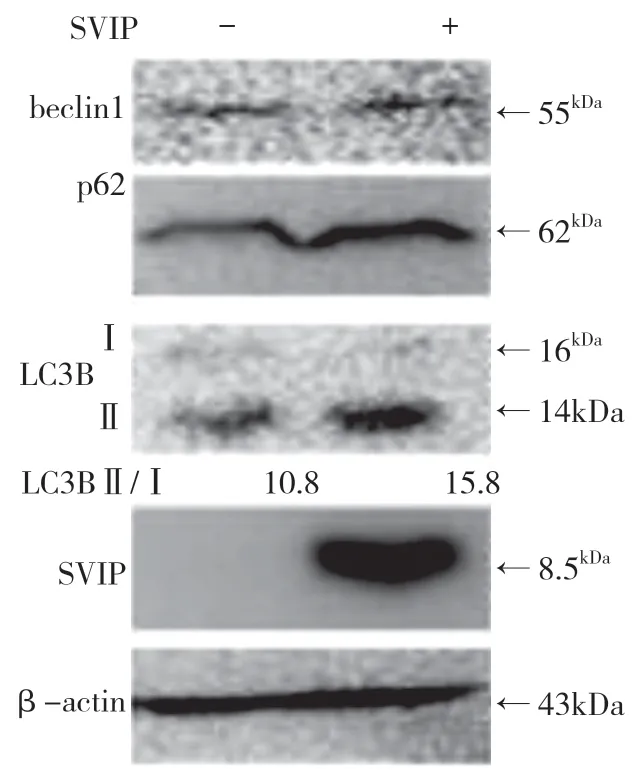
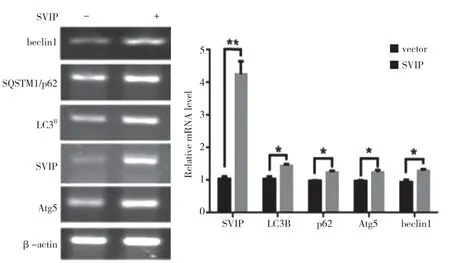
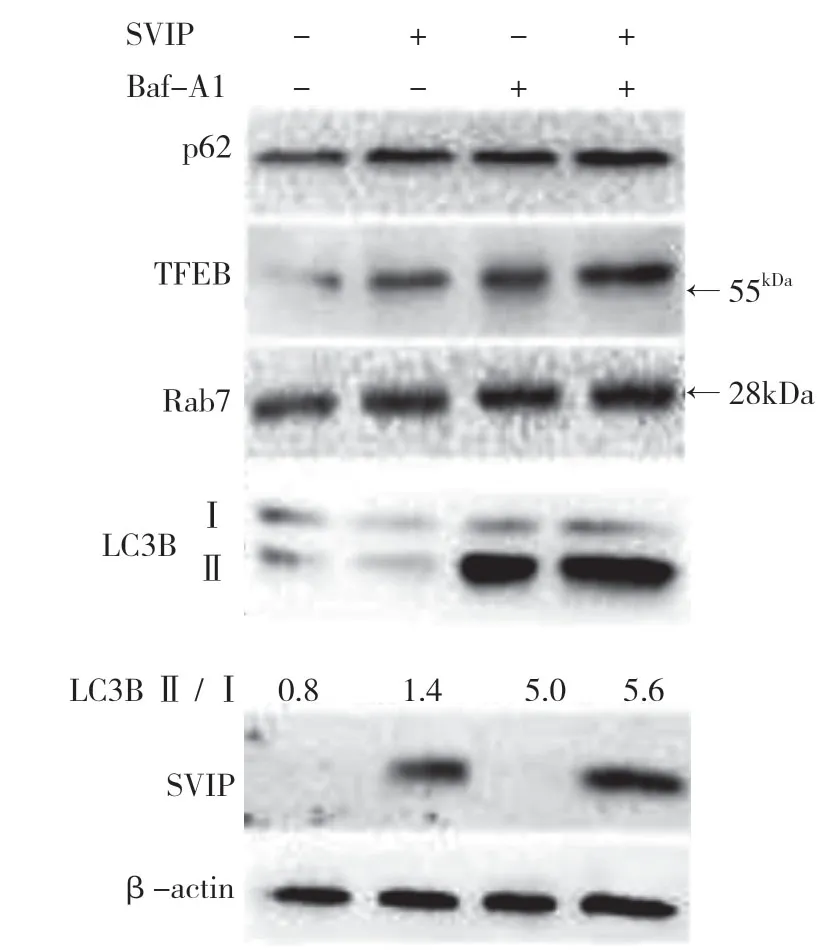
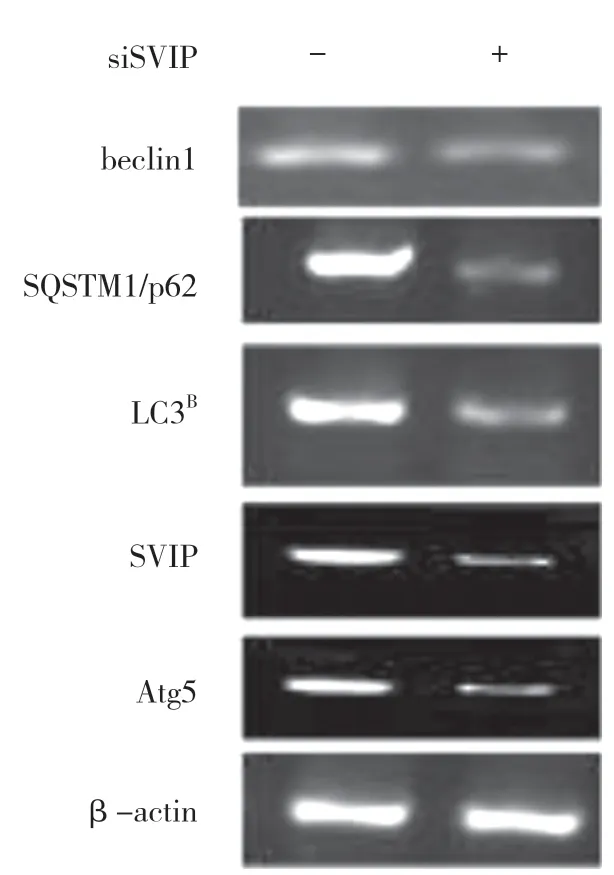
自噬相关基因的表达,包括LC3(Atg8 的哺乳动物同源物)、酵母自噬相关基因6(Atg6)、酵母自噬相关基因5(Atg5)和p62/SQSTM1,在蛋白质和基因水平上均与SVIP 过度表达呈正相关(见图a,b)。与对照组相比,它们在SVIP 细胞中的表达减少(见图e,f)。为了评估自噬通量,LC3-Ⅱ/LC3-Ⅰ通过巴非霉素A1 计算,巴非霉素A1 中和溶酶体的酸碱度并阻断自噬体-溶酶体融合(见图c,d)。转录因子EB 的表达由于SVIP 过度表达而以剂量依赖的方式增加(见图c,d),这解释了SVIP 在转录水平上增加自噬相关基因(Atgs)和p62 的表达(见图b,f)。以上所有证据表明SVIP 促进了肝癌细胞的自噬。
饥饿可以在体外和体内激活自噬。此外,SVIP 增强了饥饿诱导的自噬。但是,饥饿是否会增加肝脏或肝癌细胞中的SVIP 表达仍然未知。尽管LC3、p62、Beclin1 和Atg5 的表达因营养缺乏而显著改变,但SVIP 表达对HepG2 细胞的饥饿不敏感,也不在肝脏中,在肝脏组织中禁食24、48 h 后,SVIP 仍然是不可检测的(见图g,h,i)。同样,SVIP 表达不受小鼠大脑饥饿的影响(见图j)。
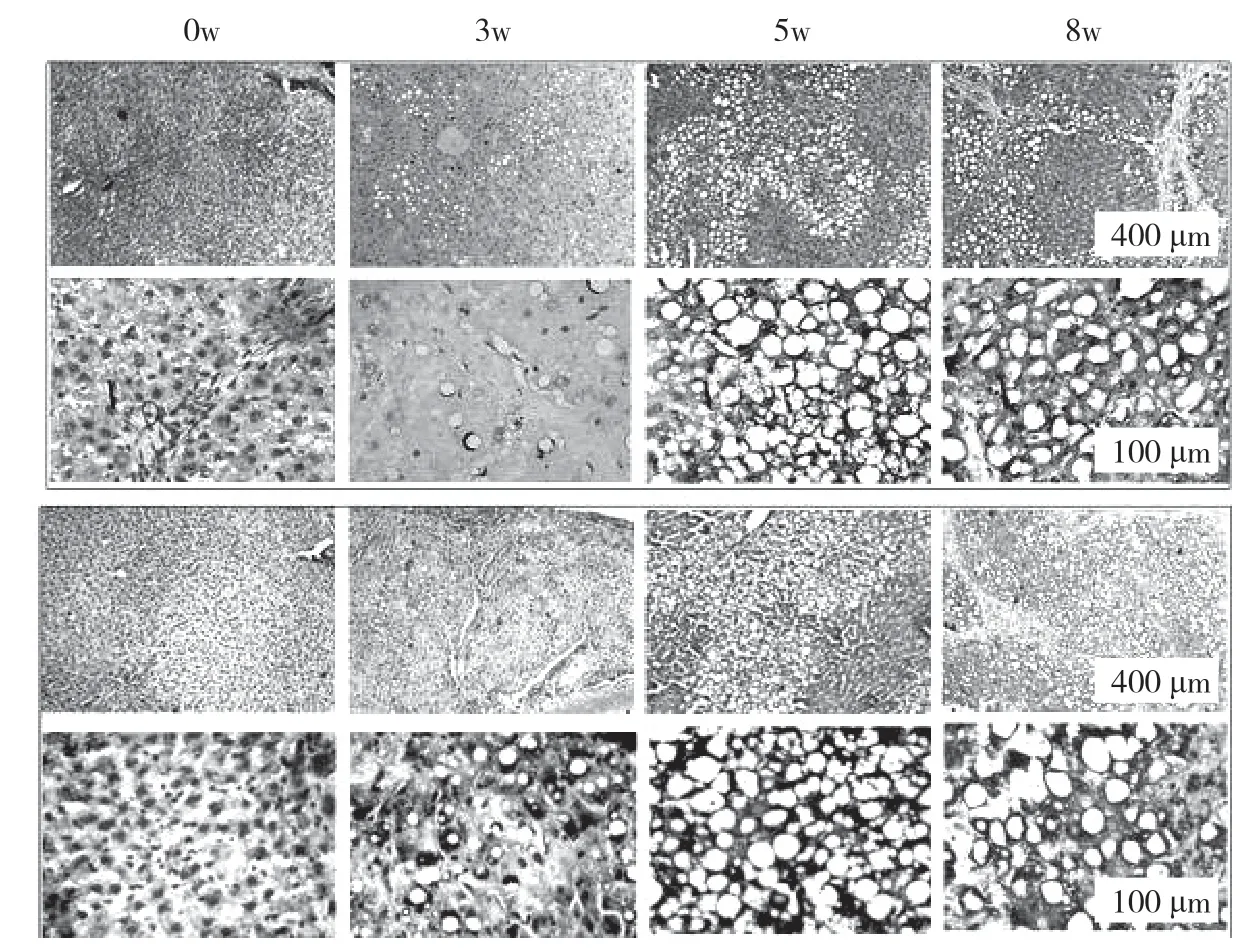
大鼠接受CCl4处理分别注射3、5、8 周。在肝纤维化的发展过程中,大鼠肝脏的病理特征发生了显著变化。最初,胶原纤维的区域和脂肪变性区域被观察到。纤维化大鼠肝脏显示不同程度的损伤:轻度,中等,严重,对应于3、5、8 周CCl4分别治疗。图表显示,倒V 形曲线以及脂滴的大小。在中度纤维化大鼠肝脏中达到顶点(见图k,l,m,n)。此外,采用抗形状记忆合金α 的天门冬氨酸氨基转移酶(AST)/丙氨酸转氨酶(ALT)生化分析测定肝功能损伤和肝纤维化的严重程度分别证实了进展中的肝纤维化,此外,分析相关基因产物的表达,蛋白质印迹试验和逆转录酶聚合酶链反应来评估自噬。SVIP 和LC3 的表达在第5 周比第3 周增加,然后在第8 周下降(见图o)。最后,SVIP 和基因水平在第3~8 周也呈倒V 型曲线。在CCl4期间治疗自噬活性在5 周达到顶峰,然后在8 周下降。这些数据表明,SVIP 的表达与自噬活性以及纤维化大鼠肝脏中脂滴的大小密切相关。
图a
图b
图c

图d

图e
图f
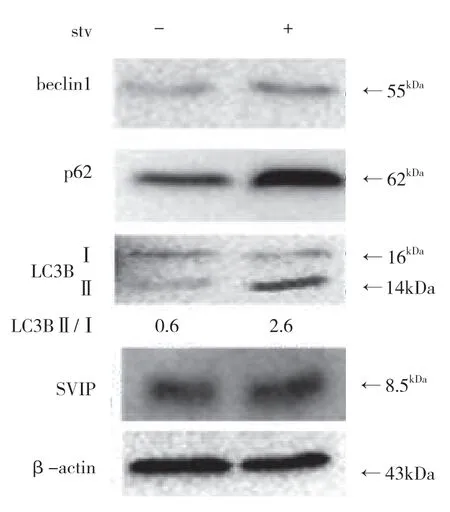
图g

图h

图i

图j

图k
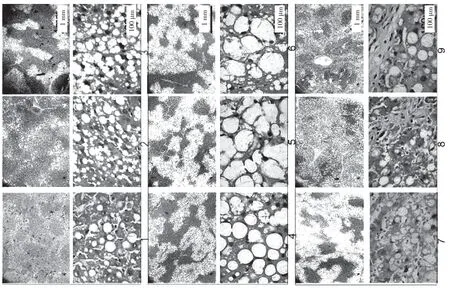
图l

图m

图n
图o
3 讨论
SVIP 保护肝细胞的机制可能包括减少脂肪变性肝脏中脂肪酸的积累和增强抗氧化作用。最初,研究表明肝细胞由于CCl4刺激导致脂肪酸的积累和脂质过氧化引起了细胞毒性。因此,SVIP 可以调节自噬,减少脂质的积累[8,9]。此外,SVIP 有一个含缬酪肽蛋白(VCP)相互作用基序,而泛素连接酶羟甲基戊二酰辅酶A 还原酶降解蛋白1(Hrd1)和STUB1/CHIP 依赖它们的VCP 结合基序降解底物。相互作用基序和结合基序都能与VCP 脱氢酶第一亚单位(ND1)结构域结合,ND1 与相互作用基序的亲和力高于VBM25。因此,SVIP 的增加竞争性地抑制了Hrd1 和STUB1/CHIP 与VCP 的结合,并减少了它们的底物降解。Nrf2 和TFEB,Hrd1 和STUB1/CHIP 的底物,是调节与抗氧化应激和溶酶体生物发生相关的基因的转录因子[10-12]。SVIP 的表达在CCl4大鼠肝脏和肝癌细胞中增多。该机制可能与泛素-蛋白酶体途径的激活有关。一些研究表明,内质网应激增加了SVIP,激活了泛素蛋白酶体途径。当CCl4导致肝细胞内质网应激,内质网相关降解和SVIP 表达增加。自噬被SVIP 激活以保护肝细胞。如果SVIP被耗尽,自噬被抑制。同时,HepG2细胞对CCl4毒性更敏感。自噬与肝脂肪变性和纤维化密切相关[13-15]。然而,从脂肪变性到纤维化过程中的自噬动力学鲜有报道。本研究结果表明自噬随着肝脂肪变性的发展而激活,然后一旦发展成纤维化就失活。这里,在CCl4中的治疗模型,激活自噬或SVIP 表达可减轻肝纤维化。随着激活的自噬,脂滴的体积不断增加,而在肝癌细胞中SVIP 过度表达或饥饿也显示出增大的脂滴。
综上所述,SVIP 的表达与CCl4形成过程中的自噬水平密切相关,且诱导肝纤维化。SVIP 能诱导自噬和延缓肝纤维化。研究SVIP 激活自噬的分子机制具有十分重要的意义,为今后抗纤维化提供了新的理论依据。
猜你喜欢
河南科技(2022年6期)2022-04-22
南方医科大学学报(2022年3期)2022-04-13
金桥(2021年10期)2021-11-05
中国医科大学学报(2021年8期)2021-09-02
浙江大学学报(农业与生命科学版)(2021年3期)2021-07-10
三农资讯半月报(2020年15期)2020-08-25
读与写(2019年35期)2019-11-05
小学生作文(低年级适用)(2019年3期)2019-04-04
中国比较医学杂志(2019年2期)2019-01-10
现代职业教育·高职高专(2018年7期)2018-05-14
