
抗肿瘤药物奥莫替尼的合成
2020-02-24 01:32:58周玲佳王秋秋朱五福
合成化学 2020年1期
肖 珍, 周玲佳, 王秋秋, 朱五福
(江西科技师范大学 药学院,江西 南昌 330013)
奥莫替尼(Olmutinib,1),化学名为N-[3-[(2-4-(4-甲基哌嗪-1-基)苯基)氨基]噻吩并[3,2-d]嘧啶-4-基]氧基]苯基]丙烯酰胺,是一种用于治疗非小细胞肺癌(NSCLC)晚期或转移性T790M突变的第三代表皮生长因子受体酪氨酸激酶抑制剂(EGFR TKI)[1-3]。该药由韩美制药开发,2016年5月获得韩国食品药品安全部(MFDS)的批准上市。1是一种不可逆激酶抑制剂,能与突变EGFR激酶结构域中的半胱氨酸残基Cys797发生共价结合作用,其半衰期超过24 h,对癌细胞株H1975(EGFRL858R/T790M)及HCC827(EGFRExon19 del)具有较强的抑制作用(GI50分别为9.2 nM和10 nM),对携带野生型EGFR的NSCLC细胞H358几乎无毒性(GI50为2225 nM)[4]。因1显著的抗肿瘤疗效,与其相关的研究受到了广泛关注[5-15]。
目前,1因为部分副作用[16]和国际药物竞争冲击而未在其他国家上市。但对1的合成工艺进行优化有助于开发新的类似小分子抑制剂。因此,本文拟对1的合成方法进行改进。

Scheme 1
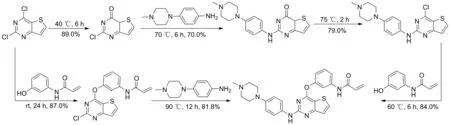
Scheme 2
文献报道的1的合成路线主要有3条。韩美制药报道的专利方法(Scheme 1)[17]:以3-氨基噻吩-2-羧酸甲酯为起始原料,经环合、氯代、亲核取代反应得到关键中间体2-氯-4-(3-硝基苯氧基) 噻吩并[3,2-d]嘧啶,再与4-(4-甲基哌嗪-1-基)苯胺进行亲核取代反应得到N-(4-(4-甲基哌嗪-1-基)苯基)-4-(3-硝基苯氧基)噻吩并[3,2-d]嘧啶-2-胺。该反应收率较低(42.0%),还原及丙烯酰化等步骤均需要使用柱层析法进行纯化。此外,硝基还原采用的是铁粉还原法,对环境污染较严重。整条路线总收率仅10%。另外两条路线皆为韩美制药对上述路线的优化。原创公司在专利[18]中报道(Scheme 2),将2,4-二氯噻吩并[3,2-d]嘧啶氧化成2-氯[3,2-d]嘧啶-4(3H)-酮后,与侧链4-(4-甲基哌嗪-1-基)苯胺通过亲核取代反应制得2-((4-(4-甲基哌嗪-1-基)苯基)氨基)噻吩并[3,2-d]嘧啶-4(3H)-酮,最后与N-(3-羟基苯基)丙烯酰胺反应得到1。该路线中对2,4-二氯噻吩并[3,2-d]嘧啶先氧化再氯代,过程较繁琐。所用中间体N-(3-羟基苯基)丙烯酰胺的合成收率较低[19]。在另一条优化路线中[20]韩美制药以2,4-二氯噻吩并[3,2-d]嘧啶与N-(3-羟基苯基)丙烯酰胺进行亲核取代反应得到N-(3-((2-氯噻吩并[3,2-d]嘧啶-4基)氧基)苯基)丙烯酰胺,再与侧链4-(4-甲基哌嗪-1-基)苯胺反应制得1。该路线的操作复杂,过程难以控制。
本文在文献基础上进行了优化,以3-氨基噻吩-2-羧酸甲酯(2)和尿素为起始原料制得噻吩并嘧啶母核,经氯代、亲核取代反应得到中间体2-氯-4-(3-硝基苯氧基)噻吩并[3,2-d]嘧啶(5),再经还原、酰胺化,最后与侧链4-(4-甲基哌嗪-1-基)苯胺通过亲核取代反应制得1(Scheme 1),总收率25.7%,其结构经1H NMR和IR确证。
1 实验部分
1.1 仪器与试剂
SGWX-4型熔点仪;ARX-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Bruker TENSOR II 型傅里叶变换红外光谱仪(KBr压片)。
所用试剂均为分析纯。
1.2 合成
(1) 噻吩并[3,2-d]嘧啶-2,4-二醇(3)的合成
将3-氨基噻吩-2-羧酸甲酯25.0 g(159.2 mmol)与脲48.0 g(792.1 mmol)加入500 mL烧瓶中,氮气保护,加热至120 ℃,搅拌下反应4 h。倒入1 L水中,冷却至室温,搅拌0.5 h,析出大量白色固体,抽滤,滤饼干燥得白色固体318.4 g,收率68.5%, m.p.138~140 ℃;1H NMRδ: 11.38(d,J=5.4 Hz, 2H), 8.04(d,J=5.2 Hz, 1H), 6.90(d,J=5.2 Hz, 1H); IRν: 3562, 1599, 1308, 1145 cm-1。
(2) 2,4-二氯噻吩并[3,2-d]嘧啶(4)的合成
将化合物318.0 g(107.0 mmol)溶解于三氯氧磷100 mL中,加入N,N-二甲基甲酰胺3 mL,搅拌下回流(120 ℃)反应3 h。反应完成之后,将反应混合物冷却至室温,缓缓加入到500 mL冰水中并剧烈搅拌,有固体析出。抽滤,滤饼用蒸馏水洗涤,真空干燥得淡灰色固体4 17.5 g,收率79.6%, m.p.121 ~129 ℃;1H NMRδ: 8.70(d,J=5.4 Hz, 1H), 7.74(d,J=5.4 Hz, 1H); IRν: 1592, 1460, 697 cm-1。
(3)5的合成
将化合物417.0 g(82.9 mmol)溶解于4-二氧六环100 mL中,依次加入间硝基苯酚12.0 g(86.3 mmol)和碳酸铯32.0 g(99.5 mmol),搅拌下反应4 h。倒入250 mL水中,搅拌10 min;抽滤,滤饼真空干燥得黄色固体525.2 g,收率98.7%, m.p.158.3~162.7 ℃;1H NMRδ: 8.51(d,J=5.4 Hz, 1H), 8.28(s, 1H), 8.16(d,J=8.2 Hz, 1H), 7.85(d,J=8.2 Hz, 1H), 7.74(t,J=8.2 Hz, 1H), 7.59(d,J=5.4 Hz, 1H); IRν: 3050, 1554, 1649, 1490, 1108, 673 cm-1。
(4) 3-[(2-氯吡啶并[3,2-d]嘧啶-4-基)氧基]苯胺(6)的合成
将化合物525.0 g(81.3 mmol)溶解于乙醇400 mL中,依次加入六水合氯化铁22.0 g(81.5 mmol)和活性炭6.8 g(566.7 mmol),升温至80 ℃,滴加水合肼40.6 g(812.0 mmol)的乙醇(100 mL)溶液,滴毕,搅拌下回流反应2 h。抽滤,滤饼用无水乙醇150 mL洗涤,收集洗液,减压蒸除溶剂,残余物加入饱和碳酸氢钠溶液200 mL,剧烈搅拌,析出固体,抽滤,滤饼干燥得黄色固体619.8 g,收率87.5%, m.p.162~163 ℃;1H NMRδ: 8.53(d,J=5.4 Hz, 1H), 7.62(d,J=5.4 Hz, 1H), 7.11(t,J=8.0 Hz, 1H), 6.53(d,J=7.8 Hz, 1H), 6.47 ~6.41(t, 2H), 5.41(s, 2H); IRν: 3448, 3122, 1542, 1319, 1181, 743 cm-1。
(5)N-{3-[(2-氯噻吩并[3,2-d]嘧啶-4-基)氧基]苯基}丙烯酰胺(7)的合成
将化合物619.8 g(71.3 mmol)溶解于二氯甲烷400 mL中,加入NaHCO312.0 g(142.9 mmol),冰浴冷却,搅拌10 min;缓慢滴加丙烯酰氯6.5 g(71.8 mmol)的二氯甲烷(10 mL)溶液,滴毕,搅拌下反应15 min。抽滤,滤饼依次用二氯甲烷和甲醇洗涤,减压蒸除溶剂,残余物用无水乙醇重结晶,抽滤,滤饼真空干燥得淡灰色固体721.2 g,收率89.6%, m.p.184~185 ℃;1H NMRδ: 10.41(s, 1H), 8.55(d,J=5.4 Hz, 1H), 7.79(s, 1H), 7.64(d,J=5.0 Hz, 1H), 7.54(d,J=8.0 Hz, 1H), 7.46(t,J=8.2 Hz, 1H), 7.10(d,J=7.0 Hz, 1H), 6.44(dd,J=16.8 Hz, 10.2 Hz, 1H), 6.27(d,J=17.2 Hz, 1H), 5.78(d,J=9.6 Hz, 1H); IRν: 3143, 1634, 1578, 1498, 1206, 695 cm-1。
(6)1的合成
将化合物721.0 g(63.3 mmol)溶解于异丙醇400 mL中,加入三氟乙酸7.3 g(64.0 mmol),升温至100 ℃,搅拌0.5 h;缓慢滴加4-(4-甲基哌嗪-1-基)苯胺14.5 g(76.0 mmol)的异丙醇(50 mL)溶液,滴毕,搅拌下回流反应4.5 h。减压蒸除溶剂,残余物加水100 mL,用稀盐酸调至pH 4,用二氯甲烷洗涤,无水硫酸钠干燥,减压回收二氯甲烷,残余物加入饱和碳酸氢钠溶液20 mL,抽滤,滤饼真空干燥得淡黄色固体118.8 g,收率60.9%, m.p. 211~212 ℃;1H NMRδ: 10.11(s, 2H), 8.55(d,J=5.2 Hz, 2H), 7.71(s, 2H), 7.64(t,J=6.4 Hz, 2H), 7.53~7.39(m, 5H), 7.04(d,J=7.0 Hz, 2H), 3.12~2.89(m, 4H), 2.38~2.29(m, 4H), 1.95(s, 3H);13C NMRδ: 174.8, 169.1, 169.3, 158.0, 149.0, 141.6, 138.2, 133.3, 131.9, 130.6, 127.9, 125.9(C2), 119.9(C2), 118.9, 117.5, 115.6(C2), 110.8, 109.0, 57.4(C2), 54.8(C2), 49.1; IRν: 3080, 1732, 1627, 1506, 1269, 1183 cm-1。
2 结果与讨论
在韩美制药的前期专利[17]路线中,合成化合物3的需要尿素6 eq.,且需先溶于N,N-二甲基甲酰胺中。反应温度高达190 ℃,反应时间为12 h。在本路线中,惰性气体保护下,直接机械搅拌原料和5 eq.尿素,反应温度降低至120 ℃,反应时间缩短至4 h。合成化合物4时,将反应温度从专利报道的200 ℃降至120 ℃;在化合物5的制备过程中,使用1,4-二氧六环替代N,N-二甲基甲酰胺作溶剂,碳酸铯用量降低至1.2 eq.。使用该路线合成时,产品收率较文献方法提高21.6%。
与韩美制药的后期专利相比,本路线详细讨论了中间体3和4的合成工艺,以及丙烯酰氯与相应中间体的反应。专利[18]采用将氯代基团先氧化后氯代的办法,总收率较低。专利[20]在合成化合物7时,产率87.0%。本路线6与丙烯酰氯进行反应,产率略微提高至89.6%。在合成1的后处理过程中,专利方法涉及抽滤、调节酸碱度、萃取和减压浓缩等一系列较繁琐的过程。本实验通过两次调节酸碱度除去了杂质,简化了后处理流程。
对抗肿瘤药奥莫替尼(1)的合成工艺进行了优化。以3-氨基噻吩-2-羧酸甲酯和尿素为起始原料,经环合制得噻吩并嘧啶母核,再经氯代、亲核取代、还原等反应合成1。与专利报道的以3-氨基噻吩-2-羧酸甲酯为起始原料的工艺路线相比,优化后的工艺路线具有后处理操作简单,总收率较高(25.7%)等优点。
猜你喜欢
证券市场周刊(2023年16期)2023-06-30 12:14:33
科技创新导报(2022年23期)2022-04-04 03:09:54
云南化工(2021年9期)2021-12-21 07:44:20
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09 07:34:10
流体机械(2020年5期)2020-06-24 05:39:08
石油钻探技术(2018年5期)2018-10-13 07:25:18
中国矿业(2017年2期)2017-02-28 02:12:56
合成化学(2015年10期)2016-01-17 08:56:44
结构化学(2014年5期)2014-12-15 08:56:20
应用化工(2014年7期)2014-08-09 09:20:26
