
蒲公英多糖脱色脱蛋白方法及其降血糖活性研究
2020-02-22 08:00郭慧静张伟达陈国刚
食品研究与开发 2020年3期
郭慧静,张伟达,陈国刚
(石河子大学食品学院,新疆石河子832000)
蒲公英(Taraxacum mongolicum)为菊科蒲公英属,是药食同源的多年生草本植物,产于黑龙江、内蒙古、山西、甘肃及新疆等省区,种质资源丰富且较易获得[1]。蒲公英除了可以鲜食以外,还可以作为药材使用,于2014 年被国家卫生计生委列入药食同源目录[2-3]。鲜食蒲公英可以起到清热解毒、消痈散结等功效;提取其功能性成分,可以有抗炎、抗肿瘤、降糖等作用;由于其药用和食用价值日益被开发,蒲公英得到了众多消费者的青睐。
植物多糖,是一种结构复杂的大分子化合物。大量研究表明从各种生物体内提取的多糖都具有多种药理活性,如抗菌、抗炎、抗肿瘤、降血糖、免疫调节和抗氧化等[4-6]功能,因此,多糖类物质的研究与开发一直是近年来的研究热点。随着科学技术的不断更新,越来越多的植物多糖被研究并加工利用。多糖也是蒲公英重要的活性成分之一,已有研究表明,蒲公英多糖具有抗菌、抗肿瘤,抗氧化等作用[7-8]。然而,多糖在提取过程中通常含有部分杂质,主要包括蛋白质和色素,不仅影响多糖的生物活性,对后期多糖的分离纯化和结构测定也带来较大困难[9]。因此,研究一种脱蛋白和脱色效果较好的工艺非常重要。
本试验以蒲公英全草为研究对象,以脱色率、脱蛋白率和多糖损失率为考察指标,对不同脱色方法和脱蛋白方法的作用效果进行比较,寻求最佳的蒲公英多糖初步纯化工艺,进一步对纯化前后蒲公英多糖的体外降血糖能力进行评价,考察其作为降血糖药物的应用前景,推动蒲公英多糖在功能食品及医药产业中的开发利用。
1 材料与方法
1.1 材料与试剂
蒲公英全草:采集于新疆哈密地区;粉末活性炭:广东韩研活性炭制造有限公司;牛血清蛋白、考马斯亮蓝 G-250:美国 Sigma 公司;木瓜蛋白酶(80 000 U/g):上海生化试剂厂;大孔树脂:北京英莱克科技发展有限公司;α-葡萄糖苷酶、对硝基苯-β-D-葡萄糖苷(4′-Nitrophenyl-beta-D-glucopyranoside,PNPG):上海宝曼生物科技有限公司;苯酚、浓硫酸、葡萄糖、无水乙醇、正丁醇、三氯甲烷、三氯乙酸等均为分析纯。
1.2 仪器与设备
HH-2 型数显恒温水浴锅:荣华仪器制造有限公司;Multifuge X1R 型高速冷冻离心机:赛默飞世尔科技有限公司;EL3002 型电子天平:梅特勒-托利多仪器有限公司;RE-3000 型旋转蒸发器:上海亚荣生化仪器厂;78-1 型磁力搅拌器:江苏金怡科技有限公司;BK-FD12T 型冷冻干燥机:山东博科科学仪器有限公司;UV-2600 型紫外分光光度计:岛津仪器有限公司。
1.3 试验方法
1.3.1 分析方法
1.3.1.1 蒲公英多糖含量测定
采用苯酚-硫酸法[10]对蒲公英多糖含量进行测定。精确称取葡萄糖标准品100 mg,用蒸馏水配成1 mg/mL的葡萄糖溶液。取上述溶液10 mL 配制成0.1 mg/mL葡萄糖储备溶液,备用。分别量取储备溶液0、0.1、0.2、0.4、0.6、0.8、1.0 mL 于试管中,加蒸馏水补至 2.0 mL,得到不同浓度的葡萄糖标准溶液。分别向上述各试管中加入6%苯酚溶液1.0 mL 摇匀,再沿壁迅速加入5.0 mL 浓硫酸混匀,沸水浴30 min 后取出冷却至室温(25 ℃),以蒸馏水作空白,于490 nm 处测吸光值。
1.3.1.2 多糖中蛋白质含量测定
采用考马斯亮蓝G-250 法[11]对多糖中蛋白质含量进行测定。精密称取100 mg 考马斯亮蓝G-250,用50 mL 95%乙醇溶解,再加入85%磷酸溶液100 mL,以蒸馏水定容于1 L 容量瓶,得考马斯亮蓝染色液。称取5 mg 牛血清蛋白溶于50 mL 0.9 %生理盐水制成0.1 mg/mL 的标准液。分别移取 0、0.2、0.4、0.6、0.8、1.0 mL 标准液于具塞试管内,用生理盐水补到1 mL,逐管加入5 mL 考马斯亮蓝G-250 染液,混匀,25 ℃室温条件下静置3 min 后,在595 nm 检测吸光值。
1.3.2 蒲公英粗多糖的制备
将采集的蒲公英清洗后50 ℃烘干,用粉碎机粉碎并过60 目筛,用石油醚回流脱脂后进行超声辅助热水提取,以1 ∶25 g/mL 料液比于75 ℃超声波清洗器中,以120 W 的超声功率浸提65 min,抽滤除去残渣,5 000 r/min 离心10 min 取上清液,将上清液浓缩至原体积的20%,加入无水乙醇使乙醇体积分数至80%,4 ℃静置过夜,4 000 r/min 离心 10 min 收集沉淀,分别用无水乙醇、甲醇、丙酮洗涤3 次,真空冷冻干燥即得蒲公英粗多糖。
1.3.3 蒲公英多糖脱色
1.3.3.1 活性炭法
取粗多糖配成5 mg/mL 粗多糖溶液20 mL,调节样液pH 值为4.0,加入2%的活性炭,在50 ℃下恒温振荡脱色50 min,按下列公式计算多糖脱色率、多糖损失率:
多糖脱色率/%=(脱色前吸光值-脱色后吸光值)×100/脱色前吸光值
多糖损失率/%=(脱色前多糖吸光值-脱色后多糖吸光值)×100/脱色前多糖吸光值
1.3.3.2 过氧化氢法
取5 mg/mL 粗多糖溶液20 mL,调节样液pH 值为9.0,加入10%的H2O2,在60 ℃恒温条件下脱色3 h,计算多糖脱色率、多糖损失率。
1.3.3.3 壳聚糖法
取5 mg/mL 粗多糖溶液20 mL,调节样液pH 值为5.0,加入5%的壳聚糖,在常温条件下振荡脱色1 h,计算多糖脱色率、多糖损失率[12]。
1.3.3.4 大孔树脂吸附法
采用 AB-8 树脂、DM301 树脂、D101 和 NKA-9 对粗多糖进行脱色处理,比较4 种大孔树脂的脱色效果。称量预处理后的树脂10 g 与20 mL 粗多糖溶液(5 mg/mL)混合,在38 ℃条件下振荡脱色2 h,抽滤,用蒸馏水洗脱,收集洗脱液,计算多糖脱色率和多糖损失率。
1.3.4 蒲公英多糖脱蛋白
1.3.4.1 盐析法[13]
取40 mL 脱色后的蒲公英多糖溶液,调节样液pH值为8,加入氯化钙粉末至10%,85 ℃搅拌10 min,冷却后离心(4 000 r/min,10 min)去沉淀,收集上清液,按下列公式计算多糖脱蛋白率和多糖损失率:
脱蛋白率/%=(脱蛋白前蛋白吸光值-脱蛋白后蛋白吸光值)/脱蛋白前蛋白吸光值×100
多糖损失率/%=(脱蛋白前多糖吸光值-脱蛋白后多糖吸光值)/脱蛋白前多糖吸光值×100
1.3.4.2 Sevag 法
取40 mL 脱色的蒲公英粗多糖溶液6 份,设置3 次~7 次脱蛋白处理。每份多糖溶液中加入1/4 体积的 Sevag(V氯仿∶V正丁醇=4 ∶1)试剂,常温振荡 30 min,离心(4 000 r/min,10 min)取上层溶液测定蛋白质含量和多糖含量,并计算多糖脱蛋白率和多糖损失率。
1.3.4.3 酶-三氯乙酸(trichloroa cetic acid,TCA)法
取粗多糖溶液40 mL,加入1.2%的木瓜蛋白酶、调节pH 为5.5,50 ℃水浴3 h 进行酶解,反应后沸水浴灭活5 min,离心取上清液。加入糖质量分数为0.1%的三氯乙酸,搅拌均匀,60 ℃水浴30 min,计算酶-三氯乙酸(trichloroacetic acid,TCA)法的脱蛋白率和多糖损失率[14]。
1.3.4.4 酶-Sevag 法
取粗多糖溶液40 mL,37 ℃水浴10 min。按1.3.4.3节酶解的条件对预热后的多糖溶液进行脱蛋白处理,对酶解后的多糖溶液按1.3.4.2 节Sevag 法再次进行脱蛋白处理,取上清液,计算酶-Sevag 法的脱蛋白率和多糖损失率[15]。
1.3.5 α-葡萄糖苷酶抑制活性测定
根据文献[16]方法并稍加修改,取浓度为0.2 U/mL α-葡萄糖苷酶溶液0.1 mL 加入2 mL 0.1 mol/L 磷酸盐缓冲液(pH 6.8),37 ℃水浴 15 min,加入样品溶液反应10 min 后再加入0.25 mL 25 mmol/L PNPG。水浴30 min后,加入0.1 mol/L 2 mL Na2CO3终止反应,于 400 nm 处测定吸光度(A2);以1 mL 缓冲液替代样品溶液,测其吸光度值为(A0);以0.1 mL 缓冲液替代酶液测定其吸光值为(A1)。重复测定3 次,并以阿卡波糖为阳性对照。
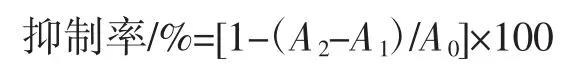
此外,酶活力定义为37 ℃、pH 6.8 条件下,1 min内水解底物(PNPG)产生1 μmol 对硝基苯酚所需的酶量为一个酶活力单位(U)。
1.4 数据分析
以上试验均在最佳条件下操作且每组数据进行3次平行试验,试验结果采用3 次重复平均值±标准差(SD)表示,采用SPSS 19.0 软件对数据进行统计分析,用Origin 8.0 软件作图。
2 结果与分析
2.1 标准曲线的绘制
葡萄糖及蛋白质的标准曲线见图1。
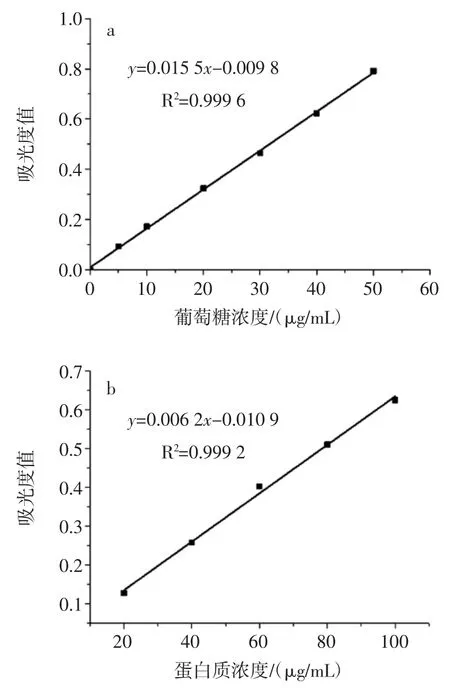
图1 葡萄糖和蛋白质标准曲线Fig.1 Standard curves of glucose and protein
以葡萄糖质量浓度为横坐标、吸光值为纵坐标绘制标准曲线如图1 a,得回归方程:Y=0.015 5X-0.009 8,式中Y 为吸光值(490 nm),X 为葡萄糖质量浓度(μg/mL),R2=0.999 6,线性范围为 0~50 μg/mL。以牛血清蛋白质量浓度为横坐标,吸光值为纵坐标绘制标准曲线如图1 b,得回归方程:Y=0.006 2X+0.010 9,式中 Y 为吸光值(595 nm),X 为蛋白质量浓度(μg/mL),R2=0.999 2,线性范围为 20~100 μg/mL。
2.2 蒲公英多糖脱色方法比较
4 种树脂的性质及对蒲公英多糖脱色效果的影响见表1。

表1 不同类型树脂的性质及脱色效果Table 1 Properties and decolorizing effect of different resins
由表1 可知,大孔树脂S-8 是极性树脂,对极性有机化合物吸附能力较强。蒲公英粗多糖溶液经S-8 树脂吸附后,脱色率达到86.45%,显著高于AB-8、DM301和D101 树脂且多糖损失率也较低,说明蒲公英粗多糖溶液中所含色素的极性与大孔树脂S-8 的极性相近,脱色效果较好。因此,选择大孔树脂S-8 作为最佳吸附介质与其它3 种方法进行比较,结果如图2 所示。
图2 多糖脱色方法的比较Fig.2 Comparison of polysaccharide decolorization methods
由图2 可知,4 种脱色方法在的脱色率均在75%以上,其中过氧化氢法脱色率最高,达到93.21%。过氧化氢是强氧化剂,可以把有色物质氧化变为无色,但其氧化色素的同时多糖也容易被分解,因此损失率相对较高。活性碳因其吸附性较强,也具有较好的脱色效果,但吸附色素的同时也易吸附一定量的多糖,使得多糖损失率相对较高。采用大孔树脂S-8 和壳聚糖进行脱色时,多糖损失率无显著差异,但大孔树脂脱色率更高,且树脂可以再生重复利用。因此,采用大孔树脂S-8 对蒲公英粗多糖溶液进行脱色。
2.3 蒲公英多糖脱蛋白方法比较
蒲公英多糖Sevag 法脱蛋白结果见表2。

表2 Sevag 法分次脱蛋白结果Table 2 The deproteinization results of polysaccharide with Sevag by different times
由表2 可知,采用Sevag 法脱蛋白时,随着试验次数的增加,蛋白质的脱除率逐渐增加,多糖损失率也逐渐增加。经6 次脱蛋白后,蛋白质脱除率趋于平缓。因此,采用Sevag 法脱蛋白时选择脱蛋白次数为6 次最佳,此时蛋白脱除率为80.82%,多糖损失率为45.16%。多糖损失较多,可能是因为Sevag 试剂在使蛋白质变性沉淀的同时会造成多糖的损失,且脱蛋白次数越多,重复操作过程也使多糖的损失增加[17]。
不同方法脱蛋白结果如图3 所示。

图3 多糖脱蛋白方法的比较Fig.3 Comparison of polysaccharide deproteinization methods
4 种脱蛋白方法的脱蛋白率均在80%以上,其中酶-TCA 法脱蛋白效果最佳,蛋白质脱除率达到90.73%,且多糖损失率仅为22.07%。然而,TCA 在水解蛋白质的同时可能会在一定程度上破坏多糖的结构,影响多糖的活性[18]。Sevag 法多次重复也可以很好地去除蛋白质,但操作繁琐耗时较多,且多糖损失率较高。酶-Sevag 法和盐析法相比,多糖损失率无显著差异(p>0.05),且脱蛋白效果均相对较好,分别为84.09%和85.64%。综合考虑脱蛋白率、多糖损失率、操作简便性及对多糖结构和活性的影响等因素,采用盐析法对蒲公英粗多糖溶液进行脱蛋白处理,进行蒲公英粗多糖的初步纯化。
2.4 α-葡萄糖苷酶抑制能力分析
蒲公英多糖对α-葡萄糖苷酶的抑制能力见图4。
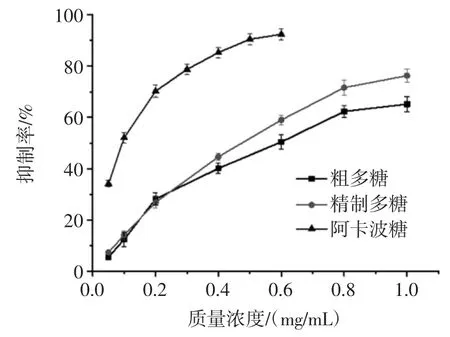
图4 蒲公英多糖对α-葡萄糖苷酶的抑制能力Fig.4 The inhibiting effects of polysaccharides from dandelion on α-glucosidase
由图4 可知,蒲公英多糖对α-葡萄糖苷酶的抑制能力随着多糖质量浓度的增加逐渐增强,呈显著的量-效关系。纯化前后蒲公英多糖对α-葡萄糖苷酶的抑制能力IC50值分别为0.65 mg/mL 和0.55 mg/mL,在质量浓度为1 mg/mL 时,抑制能力分别达到76.28%和65.16%,说明经脱色和脱蛋白后的多糖较纯化前对α-葡萄糖苷酶的抑制能力增强,可能是因为蒲公英粗多糖经处理后的纯度得到了提高。
3 结论
本试验以脱色率、脱蛋白率和多糖损失率为考察指标,比较了4 种脱色方法和4 种脱蛋白方法,结果表明大孔树脂S-8 脱色效果较好,其色素脱除率为86.45%,多糖损失率为18.9%,因此,选择S-8 树脂对粗多糖进行脱色处理。脱蛋白方法中,Sevag 法重复处理多糖损失率最高;采用酶-TCA 法处理时脱蛋白率最高,但TCA 会分解多糖从而增加多糖损失率并破坏其结构。因此,比较试验结果后选择盐析法对脱色后的蒲公英多糖进行脱蛋白处理,脱蛋白率为85.64%,多糖损失率为20.51%。蒲公英粗多糖和经初步纯化制得的精制多糖对α-葡萄糖苷酶均具有一定的抑制作用,在质量浓度为1.0 mg/mL 时,抑制率分别为65.16%和76.28%。由此可知,经本试验筛选的脱色、脱蛋白方法切实可行,降低了粗多糖中色素和蛋白质的含量,制得的蒲公英精制多糖具有良好的降血糖活性,可为进一步研究和利用蒲公英多糖提供理论依据。
猜你喜欢
农产品加工(2022年9期)2022-06-17
中国油脂(2022年1期)2022-02-12
粮食与食品工业(2021年4期)2021-08-19
作文与考试·小学低年级版(2021年12期)2021-08-09
陶瓷学报(2020年6期)2021-01-26
魅力中国(2020年8期)2020-12-07
广东农业科学(2020年9期)2020-11-10
红蜻蜓·低年级(2020年8期)2020-07-14
幼儿教育·教育教学版(2017年10期)2017-12-13
医学研究杂志(2015年12期)2015-06-10
