
DBN在蛋白质编码区识别问题中的应用研究
2020-02-18 15:20:40胡青渝刘广臣
计算机工程与应用 2020年4期
胡青渝,刘广臣
1.鲁东大学 数学与统计科学学院,山东 烟台264025
2.重庆大学 数学与统计学院,重庆401331
1 引言
目前人类基因组的全部序列已陆续测定完成,人类获取了大量的生物信息序列,并且随着外显子生物技术的不断发展,需要预测的DNA序列也在不断增加,而DNA序列中的碱基排序又十分复杂,人们想要快速又准确地获取所需要的信息十分困难。因此,如何对大量的DNA序列进行预测和分类是当代生物科学领域重要的研究课题。在过去的二十多年中,基因组序列的测定吸引了许多研究者的注意,几十种蛋白质编码区的预测方法被提出[1]。对于众多的编码区识别算法,根据其构造机理不同,可以分为两类[2-4]:依赖模型和不依赖模型。依赖模型又称为基因相似性识别算法,是以历史的基因数据作为标准,创建有标识的标准基因数据库,再根据标准基因数据库对待识别的DNA序列进行相似性识别。这种方法的准确度较高,但是对待识别的DNA序列要求十分严格。它要求待识别序列与标准数据库中的样本具有较高的同源性,而对于同源性低或者非同源的序列则无法识别。基于此,越来越多的研究者将目光转向了不依赖模型,也可称为基于统计模型的识别算法。这类方法主要建立在成熟的统计分析上,具有扎实的数据理论支撑,所获得的研究成果也远远超过依赖模型。目前较为成熟的统计模型识别算法有:Henderson和Agoes提出的隐马尔可夫模型[5]、Gelfand等提出的拼接对齐算法[6]、Howe提出的动态规划[7]、Dong等提出的语言学方法[8]、Uberbacher等提出的神经网络模型[9]、Zhang提出的线性判别分析[10]、Zhu等提出的多元熵距法[11]、Kotlar等的傅里叶分析法[12]、王飞宇等提出的基于全相位频谱分析基因识别算法[13]等。
根据已有文献可以看出,对于蛋白质编码区的识别方法虽然十分丰富,但大部分的识别算法都是建立在传统方法的基础上,它们有各自的适用对象,并且都还存在一些难以解决的问题。例如:编码区定位过界,找不到终止信息,将非编码区识别为外显子,对于较长序列的识别能力较低以及处理效率低等问题。面对当下庞大的基因数据以及不断提高的研究需求,许多算法因无法满足要求而被逐步取代。针对这个问题,2001年华盛顿大学开发了用于真核生物蛋白质编码区预测的TwinScan软件[14],它主要通过与已知的基因组序列的比较来判定待预测序列,这个方法被广泛用于哺乳动物、线虫和酵母菌等。2002年Mathe[15]提出在蛋白质编码区的识别问题中,要合理结合多种判定预测方法,对于各种方法取长补短,从而提高组合算法的识别精度。基于此,2004年Allen等[16]将动态规划法和二次判别分析等多种算法相结合,通过实验发现,该方法显著提高了识别模型的准确度。在此之后,2006年Wei和Brent[17]将TwinScan和EST相结合,提出了TwinScan_EST系统。TwinScan_EST的敏感度和特异性都优于TwinScan系统。这些结合算法使得模型对蛋白质编码区的识别更加准确,但也让实验变得更加复杂,对计算机的内存空间耗费较大,从而使得运行速度缓慢。在此基础上,提出了基于频谱分析的编码区识别算法。其关键技术是通过某些数值映射,把基因序列的碱基字符映射成数值,然后通过傅里叶变换去判断外显子所在位置。Rogic和Voss等观察发现经过傅里叶变换后的频谱序列在蛋白质编码区有明显峰值出现,在非编码区则没有这一表现[18-19]。此后,Tiwari证实了该推论,也就是蛋白质编码区存在3-周期性。
外显子的3-周期性被提出后,基于频谱的分析方法被不断提出。基于频谱分析方法的实质是通过某种数值映射方法将DNA的碱基序列映射成数值序列。2011年Sharma等对比分析了各种映射方法的构造原理[20],并对常用的12种映射方法进行了概述。其中最常见的是Voss映射,它的优点是算法的输出结果与DNA序列特征量的真实值是一致的,但是Voss映射通常用于长度为3的整数倍的序列,且效率较低,资源损耗较高。其他使用较广的方法还有Z_curve映射,它的复杂程度相对较低,但对于长度不是3的整数倍序列,它的识别效果并不理想,并且它的输出结果不能展示相应特征向量的真实值。在近几年的蛋白质编码区识别中,许多学者还提出了一些新的数值映射方法,但它们都存在各自的缺陷和弊端,如识别精度不高,识别效率较低,输出结果无法直接给出准确判断,外显子内含子识别不够准确,噪声数据算法不稳定等。
根据以上分析,可以发现目前许多结合算法依然无法做到多方兼顾,它们大多是从模型叠加或组合以及创新映射方法等角度去提高识别模型的准确度,很少从组合模型结构上进行优化。而真核生物的蛋白质编码区往往具有序列长,结构更复杂(如图1所示,不仅有编码区和非编码区,而且编码区中的外显子和内含子一般会间隔出现),编码区占比较大等特点。因此,面对庞大又复杂的DNA序列,从结构上对组合模型进行优化是十分必要的。深度置信网络则可以从结构上解决蛋白质编码区识别过程中,特征信息提取不全,分类预测不准确以及实验效率低等问题[21]。
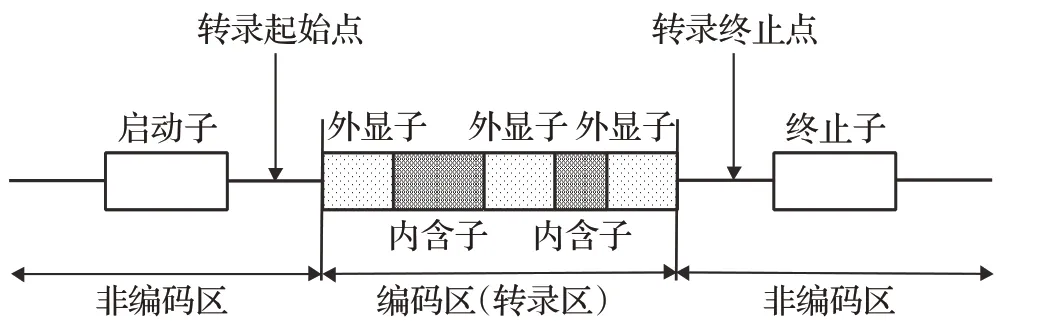
图1 真核生物的蛋白质编码区图示
深度置信网络主要通过组合底层的基础特征形成抽象的高层属性类别特征,从而发现数据的分布式特征的智能学习方法[22]。它可以根据问题的复杂程度,构建层数和每层神经元数目不同的网络模型,它的实质是通过大量的训练数据和构建多层限玻尔兹曼机来学习有用的特征,从而提升模型的分类和预测能力。深度置信网络强调了模型结构的深度,突出了特征学习的重要性,通过逐层特征变换,将样本在原有空间中的特征变换到一个新的特征空间,从而使得分类预测更加容易,同时还提高了分类预测的效率[23]。与传统的仅使用人工构造的规则来提取编码区特征的方法相比,深度置信网络利用大数据来训练模型,从而获得编码区特征,该特征能够更详细准确地刻画数据的丰富内在信息。
基于此,本文提出了可以综合多种数值映射方法,并且能够更加准确地判别预测和定位蛋白质编码区的组合识别模型。它主要通过多层玻尔兹曼机进行特征降维,并提取影响编码区识别的关键因素,并在深度置信网络最后一层加上神经网络判别分类器,实现对蛋白质编码区的判别和预测。通过实证表明,基于深度置信网络的非监督方法能够在一定程度上提高蛋白质编码区识别问题的预测精度,提高组合模型的有效性和实用性,同时还大大降低了训练模型的复杂程度。这对于真核生物蛋白质编码区的识别问题具有十分重要的意义。
综上所述,本文的主要工作是对复杂的真核生物蛋白质编码区结构中的外显子和内含子进行判别和定位,并且充分利用深度置信网络的结构优势来提高识别模型的准确度和实验效率。模型的具体构造过程如下:
(1)利用信号转换算法将复杂的DNA字符串转变为数值序列,也就是将四种碱基转换成数值形式,然后再结合基础统计学方法对这些数值序列进行特征提取。
(2)由于特征数量众多,并且某些特征存在相关性,利用随机森林的方法对众多特征进行变量选择。
(3)将提取出的特征集作为判别变量,已知的编码区判别结果作为判别目标来构建深度置信网络模型,最后将经过数值转换的DNA序列分为训练集和测试集对模型进行训练和测试。
2 数据的采集与预处理
2.1 数据的采集
对于蛋白质编码区的识别问题,本文主要是对真核生物的DNA序列进行判别分析,因此使用的数据也是真核生物的DNA序列,包括BG570、HMR195以及GENSCAN65数据。数据的主要来源是BG570数据集(http://www1.imim.es/databases/genomics96/),HMR195数据集(http://www.cs.ubc.ca/~rogic/evaluation/),GENSCAN65数据集(http://www.ncbi.nlm.nih.gov/nuccore/FO081497),这里统称它们为原始数据。因为原始数据中有部分DNA序列是小于20bp的,这些数据包含的信息是不全面的,所以本文首先将这些数据从数据集中剔除,然后从原始数据中选取长度为20bp以上的外显子和内含子。新建的基本数据集中外显子和内含子的个数如表1所示。

表1 剔除信息缺失序列后的外显子和内含子数据分布表
2.2 数据的预处理
为了能够使深度学习算法对真核生物蛋白质编码区进行分类和预测,首先需要对DNA序列进行数值化映射,也就是将DNA字符序列转换为数值序列,然后再提取这些数值序列的数字特征,最后将这些数值序列作为训练数据输入到深度置信网络模型中对DNA序列进行分类训练。
近年来,DNA谱分析技术已被引入蛋白质编码区识别的研究中,这类算法主要利用数字信号处理技术,通过计算比较蛋白质编码区和非编码区的功率谱密度信噪比曲线的特征,例如三周期特性等差异来进行编码区识别。这类方法并没有利用传统的序列对比思想,如BLAST,因此称它为非序列比对特征提取。非序列比对特征提取主要包括两个阶段:一是对DNA序列进行数值化映射;二是提取DNA数值序列的特征。
2.2.1 DNA序列的数值化映射
本文主要采用信号处理技术对DNA序列进行数值转换。本文所采用的信号处理技术[24-25]主要可以分为两种类型:第一类是K字符相对频率技术,K字符指的是DNA序列中长度为K的连续核苷酸片段,例如K=1表示核苷酸A、C、G、T;K=2是指AA,AC,…,TT,以此类推。第二类是重编码技术,包括8种固定映射技术[26-27],7种基于物理化学性质的映射方法[28-29],4种基于DNA图表达的长程相关性方法[30]。映射公式如表2~表4所示。

表2 固定映射技术公式表

表3 基于物理化学性质的映射方法公式表

表4 基于DNA图表达的长程相关性方法公式表
2.2.2 DNA序列的特征提取
本文通过2.2.1小节将DNA字符序列转换为数值序列,若要实现深度置信网络模型对蛋白质编码区的分类预测,还需要从这些数值序列中提取出数字特征,进而构成训练集和测试集。下面着重介绍本文所采用的特征提取方法以及所提取出的数字特征[31]。
(1)基于K-tuple的数值特征提取
以K=2时GT的相对丰度为例,进行数值特征的提取:

其中,fG、fT、fGT分别代表核苷酸G、T和双核苷酸GT在基因片段中的频率。相似的,可以根据相对丰度的公式及其推广来计算其他情况下的相对丰度。对于较长的DNA序列,提取出的K-tuple数量将自动地随序列数量的变化而变化,即由min(Si)来确定,这将决定不同长度的序列所提取的数值特征的准确性及最终预测的准确性。
综上所述,利用相对丰度可以提取到85个数值特征。
(2)基于重编码的数值特征提取
根据不同的重编码技术,可以获得不同的数值映射结果,具体方法已在数值化映射中做出了阐述,对于这些不同的重编码技术获得的数值序列,将采用计算各阶矩的方法进行特征提取。根据重编码器获得的数值特征可以分为两类:一类是实数类重编码技术的数值特征提取;另一类是复数类重编码技术的数值特征提取。
对于实数类重编码技术的数值特征提取,首先是Voss方法,它具有良好的特征表达作用,这也使它成为了基因组谱分析中最著名的数值映射技术。对于Voss变换后的数值序列,通过计算其一、二、三阶矩的方法来提取数值统计特征,总共得到了12个特征。其次对于实数类序列,计算其他常用特征,如均值、标准差、偏度值和峰度值,并得到了88个特征。最后通过计算各实数序列的Hurst指数来对数据的特征进行提取。Hurst指数是英国水文学者Hurst在研究尼罗河水文时基于R/S法提出的一种用来刻画时间序列相关性的指标,后来成功被引入来比较DNA序列的相似性。利用该方法可以获得24个特征。
对于复数类重编码技术的数值特征提取,经过快速傅里叶变换得来的复数序列,可以计算其傅里叶系数的平方值序列,然后计算其功率谱均值,这样一共可以得到28个特征。
综上所述,利用重编码方法一共可以获得152个特征。而经过计算K-tuple的相对丰度和重编码计算的各类数值特征提取,一条无论长度为多少的DNA序列都可以用一条长度为85+152=237的数值特征序列来代替,不过这237类数值特征可能存在共线性或对编码区识别的不显著性,因此下文将会用到随机森林的方法来消除具有共线性和不显著性的特征变量。本例共有7 081条不同长度的DNA序列,经过上述转换和特征提取后,将会得到7081×237的数字矩阵,称为原始特征矩阵,矩阵的行向量表示原始的DNA序列,列向量表示每条DNA序列的一个数值特征。具体矩阵如表5所示。
2.3 基于随机森林的特征子集选择
根据上文可知,经过特征提取一共获得了152个特征变量,而这些变量中不乏许多噪音,也就是对DNA序列的判别没有显著性关系的特征,这些特征非但不能帮助模型识别蛋白质编码区,反而会干扰模型的判别能力,因此考虑对152个特征进行降维。由于这些数值特征是离散的,并且无法通过理论分析判断它们与模型识别效果的相关程度与重要性,本文考虑利用随机森林对特征变量进行降维选取。随机森林是一种集成机器学习算法[31-33],它可以利用随机重采样技术(bootstrap)和节点随机分裂技术构建一片由决策树组成的森林,并且让这片森林里的所有决策树都参与投票,计算出每一个特征变量的重要程度值,最后根据计算所得的重要性对这些特征进行排序。相较于其他降维方法,随机森林在对变量进行选择时具有以下优势:

表5 DNA数值特征表
(1)对于DNA序列这种变量数据集较多的数据精确度较高。
(2)不易发生过拟合的现象,这对于模型在测试集上的预测效果有重要的意义。
(3)可以处理离散化的数据,因此对于种类众多的基因特征数据,无需进行归一化处理就可以选出与DNA判别具有显著关系的变量特征。
(4)具有良好的处理缺失数据的能力,由于使用的DNA序列集中序列长度并不是一定相等的,就算去掉长度小于20bp的序列,仍会在特征提取时存在特征数据不全的情况。而随机森林这一特性则很好地解决了这个问题。
(5)最重要的一点是它不但可以对特征变量进行降维,还可以得到特征变量的重要性排序,这对人们选择进入深度置信网络判别模型的初始特征变量提供了十分重要的理论依据。
(6)具有以上优点的随机森林的实现并不复杂,且容易并行化,这在构建组合模型时大大提高了模型的判别和预测效率。
基于此,本文利用R语言中的RandomForest包实现了特征变量的提取,计算出了每个特征变量的得分,并根据其重要性排序选出了前50%的变量,即119个变量,放入判别模型中。
图2展示了得分较高的30个特征变量,其中得分最高的特征变量为V 129,最后选定了前50%的特征变量放入深度置信模型中,实现了对特征变量的降维。利用随机森林对特征变量进行降维,首先可以剔除无关变量或者与模型显著性关系较小的变量,降低由无关特征变量引起的噪声对模型判别效果的影响;其次提升了特征变量的可解释性,并且特征变量的减少还提高了组合模型的实验效率;最后将处理后的特征变量作为初始变量放入深度置信网络中,可以使模型更有效地习得识别蛋白质编码区的方法,从而提高模型的判别和预测能力。
2.4 确定训练集和测试集数据

图2 部分重要变量得分曲线图
为了更好地对深度置信模型进行训练,将原始数据集分为两部分,一部分作为训练集,另一部分作为测试集,其中前70%的数据作为测试集,后30%的数据作为训练集。具体数据分布如表6所示。

表6 训练集和测试集数据分布表
3 真核生物蛋白质编码区识别模型的建立3.1 构建深度置信网络模型
深度置信网络(Deep Belief Network,DBN)是深度学习方法中的一种常用模型[34-35],也是神经网络的一种。深度置信网络既可以用于非监督学习,将它看作一个自动编码器;也可以用于监督学习,将它看作一个分类器。本文主要将深度置信网络作为分类器来使用,首先通过无监督学习框架得出特征向量,然后再将特征向量赋给神经网络(Artificial Neural Network,ANN)模型完成分类。
深度置信网络是由多个限波尔兹曼机(Restricted Boltzmann Machine,RBM)模型构成的。RBM是由神经网络所衍生出的一种感知器,主要由显层和隐层两部分构成,其中显层和隐层的神经元为双向链接。任意两个相连神经元之间的链接强度由权重W表示;对于每一个显层神经元都有一个偏置系数b用来表示它的自身权重;对于每一个隐层神经元也都有一个偏置系数c用来表示它的自身权重。
RBM的能量函数为:

其中,h表示隐层神经元(hidden),v表示显层神经元(visible)。
RBM中隐层神经元被显层神经元激活的概率函数为:

RBM中显层神经元被隐层神经元激活的概率函数为:

因为同层神经元之间是相互独立的,所以它们的概率密度也是相互独立的,由此可以得到:

当给显层神经元输入一列数据后,RBM可以根据式(3)计算出每个隐层神经元被激活的概率P(hj|x),j=1,2,…,Nh,取阀值μ为0~1的随机数,概率大于该阀值的隐层神经元被激活,否则不被激活,判别式为:

由此可以判断隐层的每个神经元是否被激活。若赋值给隐层,显层的神经元是否被激活的计算方法也是一样的。
3.2 对限波尔兹曼机(RBM)模型的训练
RBM中共有5个参数h、v、b、c、W,其中v是输入向量,h是输出向量,b、c、W是相应的权重和偏置值,是通过数据学习得到的。对于一系列样本数据x,主要采用对比散度的算法进行训练[36]。
(1)将一系列数据x赋给显层v1,然后利用式(3)计算出每个隐层神经元被激活的概率P(hj|x),j=1,2,…,Nh;
(2)从这些计算得出的激活概率分布中采用Gibbs方法抽取一个样本集:h1~P(h1|v1);
(3)利用隐层神经元h1重构显层,即通过隐层反推显层,可以通过式(4)计算出显层中每个神经元被激活的概率:P(v2|h1);
(4)从计算得到的激活概率分布中再利用Gibbs抽样法抽取一个样本集:v2~P(v2|h1);
(5)通过v2再次计算每个隐层神经元被激活的概率,得到的概率分布为:P(v2|h1);
(6)更新权重:

经过反复的训练后,隐层神经元不但能较为准确地显示出显层神经元所包含的特征,并且还能够还原显层信息。当隐层神经元数量小于显层神经元数量时,就会起到“压缩数据”的效果。
最后将若干个RBM“串联”起来就能构成一个深度置信网络,其中上一个RBM的输出层也就是下一个RBM的输入层,上一个RBM的隐层即为下一个RBM的显层。在整个训练过程中,对上一层的RBM进行充分训练后才能接着训练当前层的RBM,直到最后一层。
3.3 深度置信网络模型的求解
3.3.1 分类结果及分析
对于模型的实现本文采用加拿大多伦多大学Ruslan Salakhutdinov和Geoff Hinton的软件包[35-36],利用训练集对模型进行训练,然后利用测试集对模型进行了误差分析。本文所用的数据集一共包含7 081个样本,需要判别的类型有外显子和内含子两类。本文将70%的数据作为训练集,30%的数据作为测试集。
首先利用训练集对深度置信网络模型进行无监督训练。本文所构建的深度置信模型由4个RBM模型“串联”而成,因此模型分为4层,根据Kolmogorov定理确定每层所含隐藏神经元个数分别为119,50,50,200。本文列出了深度置信网络里每层神经元的输出结果,也就是每层神经输出的特征向量。具体结果如表7所示。
最后将深度置信网络无监督学习得来的特征向量放入深度置信网络的分类器中,本文所用的分类器是神经网络分类器,由此可以得到蛋白质编码区的预测结果。根据放入测试集的分类标签可知,当预测结果为1时,该测试序列被模型判别为外显子,当预测结果为2时,该测试序列被模型判别为内含子。对比DNA序列原有的分类标签,可以得出本文提出的深度置信网络模型的准确率为83.43%。
3.3.2 对比分析
为了进一步验证深度置信网络对蛋白质编码区识别问题的准确度,还运用了传统的Logistic回归分类器和贝叶斯判别法对蛋白质编码区进行了判别分析。具体预测结果分析如表8所示,其中正确率为外显子被正确预测的比率;灵敏度为所有实际外显子中被正确预测为外显子的比例;精确率为预测为外显子的序列中真正为外显子的比例;特异度为所有真实的内含子序列被正确预测为内含子的比例。由于灵敏度和精确率两个指标有时会出现方向不同的结果,因此引入F得分,即灵敏度和精确率加权后的调和平均。根据以上五种评价指标[31]可以看出,相比于利用传统的Logistic回归和贝叶斯判别法对蛋白质编码区进行识别,本文所使用的深度置信网络对蛋白质编码区具有更好的识别功能,也就是说,本文所使用的深度置信网络模型能够从众多的DNA序列数据中提取出有效的信息对外显子和内含子进行识别。
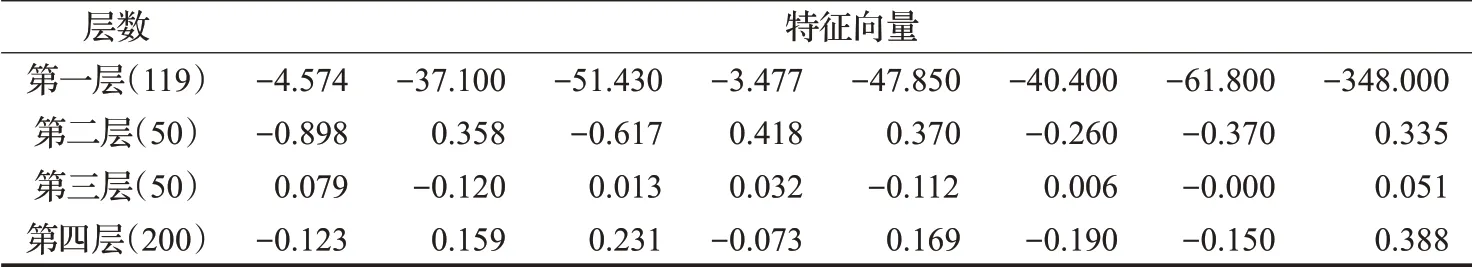
表7 DBN每层特征向量的输出结果展示

表8 三类判别分析的结果分析指标表 %
3.4 预测结果分析
为了更加准确地判断组合模型的识别效果,本文以测试DNA序列的前5条为例,由于每条测试序列中既包含编码区也包含非编码区,且它们一般是无规律间断出现的,本文采用了一个移动平滑框对序列进行截取。图3是移动平滑框截取DNA序列的模拟图。

图3 移动平滑框截取DNA序列的模拟图
如图3所示,数据框沿着测试DNA序列滑动,位于数据框结尾位置的碱基为C,数据框中包含有碱基C及其上游的一共250个碱基。如果被截取的序列被模型判定为外显子,则说明碱基C之前的250个碱基构成的序列被判定为外显子,此时模型的输出值为“1”。然后数据框将沿着DNA序列向前移动一个碱基,此时数据框结尾处的碱基为G,若此时模型的输出值为“1”则说明碱基G也为外显子,若模型的输出值为“2”则说明碱基G被模型判定为内含子中的碱基。以此类推,一条长度为N的DNA序列可以被切分成N-249条测试序列放入模型中进行判别,从而确定该条序列中外显子和内含子的具体位置和数量。本文所采用的5条测试序列长度分别为2 176、4 775、13 054、7 658、3 967。为了更清晰地展示本文所提出的模型对蛋白质编码区的预测效果,利用混合矩阵将预测值和真实值进行比较,结果如表9~表13所示,其中1表示外显子,2表示内含子。

表9 第一条DNA序列混淆矩阵对外显子识别结果

表10 第二条DNA序列混淆矩阵对外显子识别结果

表11 第三条DNA序列混淆矩阵对外显子识别结果

表12 第四条DNA序列混淆矩阵对外显子识别结果

表13 第五条DNA序列混淆矩阵对外显子识别结果
通过查看表9~表13对角线上的数据可以看出,在DNA测试序列上本文所提出的组合模型的判别效果是比较准确的,被错误判断的序列占比较小。
为了更加直观地评估模型的性能,在混淆矩阵的基础上分别计算了5条测试序列的准确率、灵敏度、精确率、特异度以及F得分。具体结果如表14所示。

表14 5种评价指标得分表 %
根据表14的5种指标可以看出,利用移动平滑框结合深度置信网络对DNA序列中的蛋白质编码区进行预测是可行的,并且准确率较高。并且对于不同的DNA序列,模型识别的精确程度有小范围的不同,但准确率都基本维持在98.48%左右。评测了模型的预测效果后,考虑如何对内外显子进行准确定位,本文主要采用了短时傅里叶变换技术(Short Time Fourier Transform,STFT)对编码区进行准确定位。
最后,为了更清晰地看出模型所预测的蛋白质编码区所在区域位点,本文采用了STFT定位技术来对DNA测试序列的深度置信网络判别模型输出值进行分析。STFT是一种常见的时频分析方法,通过一个时间窗口内的一段信号来表示某一时刻的信号特征,也就是把深度置信网络模型的输出值和窗函数相乘,然后进行一维傅里叶变换,再通过窗函数的滑动得到一系列的频谱值,将这些结果映射到坐标轴上便得到一个二维的时频图。结果发现外显子和内含子通过DBN输出的频谱值差别是很大的,那么找到频谱出现变化的起始点,也就找到了蛋白质编码区,即外显子的确切位置。
本文以测试集的前5条DNA序列为例,将深度置信网络模型的输出值输入STFT模型中对编码区进行定位,由此可以得到不同DNA序列的时频谱在位置轴上的投影,如图4所示。从图中可以看出,外显子区域和内含子区域在固定频率上是不一样的,也就是说在位置轴上投影的幅频特性有明显区别。因此根据DNA序列的STFT时频分析,可以确定出编码区的位置,其中峰值为2的是内含子所在区域,峰值为1的为外显子所在区域。

图4 5条测试DNA的时域波形图
根据图4可以看出,蓝线代表DNA序列的真实位点,红线代表DNA序列的预测位点,它们时域图重合的部分表明模型正确预测内外显子的位点,反之则预测有误。从图中可以看出,本文提出的基于深度置信网络的组合识别模型的判别能力较强,预测的位点也比较准确。
4 总结
本文着重探讨了如何利用生物统计学方法从真核生物DNA序列中识别出蛋白质编码区域,主要阐述了对DNA序列的数值转换、特征变量的提取、特征变量的降维以及对DNA序列的分类预测和对蛋白质编码区的准确定位。本文涉及了多种生物统计学方法,其中包括信号处理技术、重编码技术、随机森林、深度置信网络以及STFT定位技术。将深度置信网络模型运用到蛋白质编码区的识别问题,突破了传统蛋白质编码区识别技术的壁垒。为了更好地证明模型的可行性,本文还利用了Logistic回归模型和贝叶斯判别模型与深度置信网络模型进行了对比,最后发现基于深度置信网络模型的蛋白质编码区识别技术在各项指标的评定下具有更好的实证效果,这对于生物信息学的研究起着十分重要的作用。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
自然杂志(2021年6期)2021-12-23 08:24:46
中国毕业后医学教育(2021年3期)2021-12-02 02:24:20
中国毕业后医学教育(2021年3期)2021-12-02 02:24:18
陶瓷学报(2021年2期)2021-07-21 08:34:58
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
现代装饰(2018年5期)2018-05-26 09:09:01
华东理工大学学报(自然科学版)(2015年2期)2015-11-07 09:16:29
电源技术(2015年5期)2015-08-22 11:18:38
