
酚氨咖敏颗粒质量标准研究
2020-02-11 07:59董秋香张月寒付萍萍
中国药业 2020年3期
董秋香,张月寒,姚 辉,付萍萍,王 腾,王 楠
(河北省保定市食品药品检验所,河北 保定 071051)
酚氨咖敏颗粒曾用名为克感敏颗粒、克感敏冲剂,是解热镇痛类处方药,用于治疗感冒、发热、头痛、神经痛及风湿痛等疾病。该药由对乙酰氨基酚、氨基比林、咖啡因及马来酸氯苯那敏4种成分组成,现行标准[1]中缺少主要成分对乙酰氨基酚的鉴别及含量测定,其他成分的含量选用2种不同的高效液相色谱(HPLC)系统分别测定。酚氨咖敏片现行标准[2]采用HPLC法测定4种成分的含量,因各成分处方量及其紫外吸收强度差异均较大,需分别制备2种供试品溶液在不同波长下测定,无法实现同时检测。郭琦等[3]报道,采用气相色谱(GC)内标法可同时测定4种成分。为加强对该药品的质量控制并简化操作,本研究中建立了快速定性鉴别4种成分的薄层色谱(TLC)法,以及可同时测定4种成分含量的GC外标法。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 7820A型气相色谱仪,包括FID检测器,G4513A型自动进样器,EC Chrom Elite工作站(美国Agilent公司);Sartorius BP211D型电子分析天平(德国Sartorius公司,十万分之一);CAMAG型薄层色谱成像系统(瑞士Camag公司);SK250HP型超声波清洗器(上海科导超声仪器有限公司)。
1.2 试药
酚氨咖敏颗粒(市售品,批号分别为16062211,15121471,15121375);对乙酰氨基酚对照品(批号为100018-201610,纯度为99.9%),氨基比林对照品(批号为100503-201302,纯度为99.9%),咖啡因对照品(批号为171215-201512,纯度为99.9%),马来酸氯苯那敏对照品(批号为100047-201507,纯度为99.7%),均购自中国食品药品检定研究院;硅胶GF254板(德国Merck公司);其他试剂均为分析纯。
2 方法与结果
2.1 薄层色谱鉴别
称取对乙酰氨基酚、氨基比林、咖啡因、马来酸氯苯那敏对照品适量,加三氯甲烷制成质量浓度分别为20,25,6,0.4 mg/mL的混合溶液,必要时滤过,取滤液作为对照品溶液。称取本品细粉适量(约相当于马来酸氯苯那敏4 mg),加三氯甲烷10 mL,振摇使溶解,滤过,取滤液作为供试品溶液。除去4种待测成分,模拟处方(生产企业提供)按供试品溶液制备方法制备阴性对照品溶液。分别吸取上述3种溶液各10μL,点于同一硅胶GF254板上,三氯甲烷-甲醇-丙酮-乙醚-氨水(9∶0.8∶1∶5∶0.2,V/V/V/V/V)为展开剂,展开(展开距离约为8 cm),取出,晾干,置紫外光灯(254 nm)下检视。紫外光灯下,4种待测成分完全分离,比移值分别为0.1,0.4,0.5,0.8,3批样品所显斑点颜色与位置分别与对照品相同,辅料无干扰。色谱图见图1。

图1 薄层色谱图
2.2 含量测定(GC法)
2.2.1 色谱条件
色谱柱:TR-1弹性石英毛细管柱(30 m×0.32 mm,0.25μm,S/N:11019B03,Thermo科技公司);升温程序:起始温度为180℃,保持1 min,再以8℃/min的速率升至240℃,保持2 min;进样口温度:240℃;分流比:20∶1;载气:氮气;柱流量:1.5 mL/min;检测器:FID,温度260℃;氢气流量:30 mL/min;空气流量:400 mL/min;尾吹流量:25 mL/min;进样量:1μL。

图2 气相色谱图
2.2.2 溶液制备
对照品溶液:取对乙酰氨基酚、氨基比林、咖啡因、马来酸氯苯那敏对照品适量,精密称定,加乙醇制成质量浓度分别为15.0,10.0,3.0,0.2 mg/mL的混合溶液,作为对照品贮备液;精密量取对照品贮备液5 mL,置50 mL容量瓶中,加乙醇稀释至刻度,摇匀,即得。
供试品溶液:取本品10包,精密称定内容物总质量,研细,取细粉适量(约相当于马来酸氯苯那敏1 mg),精密称定,置100 mL具塞锥形瓶中,精密加入乙醇50 mL,超声(功率250 W,频率53 kHz)提取20 min,放冷,滤过,取续滤液,即得。
阴性对照品溶液:除去4种待测成分,以模拟处方按供试品溶液制备方法制备。
2.2.3 方法学考察
专属性试验:取2.2.2项下3种溶液各1μL,按拟订色谱条件进样测定,记录色谱图,详见图2。结果,在此色谱条件下,4种待测成分峰与相邻峰分离良好,分离度均大于1.5,辅料对测定无干扰。
线性关系考察:精密量取对照品贮备液适量,加流动相等倍逐级稀释成系列质量浓度的对照品溶液,按拟订色谱条件进样测定,记录峰面积。分别以4种待测成分的质量浓度(X,μg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,对乙酰氨基酚、氨基比林、咖啡因和马来酸氯苯那敏的回归方程分别为Y乙=3.999×103X乙-1.238×105(r=0.999 9),Y氨=4.506×103X氨+3.736×104(r=0.999 9),Y咖=2.391×106X咖-2.125×103(r=1.000 0)和Y马=3.922×106X马+5.964×103(r=0.999 9)。结果表明,该4种成分质量浓度分别在186.3~596 0μg/mL,124.9~3 996μg/mL,38.24~1 224μg/mL,2.525~80.80μg/mL范围内与峰面积线性关系良好。
定量限(LOQ)测定:取2.2.2项下的对照品贮备液适量,等倍逐级稀释,按拟订色谱条件进样测定。结果对乙酰氨基酚、氨基比林、咖啡因和马来酸氯苯那敏的LOQ分别为0.09,0.06,0.10,0.07 ng。
精密度试验:取2.2.2项下混合对照品溶液适量,按拟订色谱条件进样测定,记录峰面积。结果对乙酰氨基酚、咖啡因、氨基比林和马来酸氯苯那敏峰面积的RSD分别为0.69%,0.50%,0.65%,0.68%(n=6),表明仪器精密度良好。
稳定性试验:取样品(批号为15121471),依法制备供试品溶液,分别于室温下放置0,1,2,4,6,8,10,12,24 h时,按拟订色谱条件进样测定,记录峰面积。结果对乙酰氨基酚、氨基比林、咖啡因和马来酸氯苯那敏色谱峰面积的RSD分别为1.69%,1.78%,1.75%,1.29%(n=9),表明供试品溶液在室温下24 h内稳定。
重复性试验:取同一批(批号为15121471)样品,依法制备供试品溶液6份,按拟订色谱条件进样测定,以外标法计算平均含量。结果样品中对乙酰氨基酚、氨基比林、咖啡因和马来酸氯苯那敏的平均含量分别为99.51%,99.53%,99.32%,98.48%,RSD分 别 为0.58%,0.70%,1.79%,1.15%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的样品(批号为15121471),取细粉适量(约相当于马来酸氯苯那敏1 mg)9份,精密称定,置100 mL具塞锥形瓶中,分别精密加入低、中、高质量浓度的混合对照品溶液,依法制备供试品溶液(n=3),按拟订色谱条件进样测定,计算加样回收率。结果见表1。
2.2.4 含量测定
取3批样品,依法制备供试品溶液,按拟订色谱条件进样测定,记录峰面积,以外标法计算4种成分的含量,咖啡因计算结果与1.092 7相乘,并与现行标准检验结果进行比较。结果见表2。
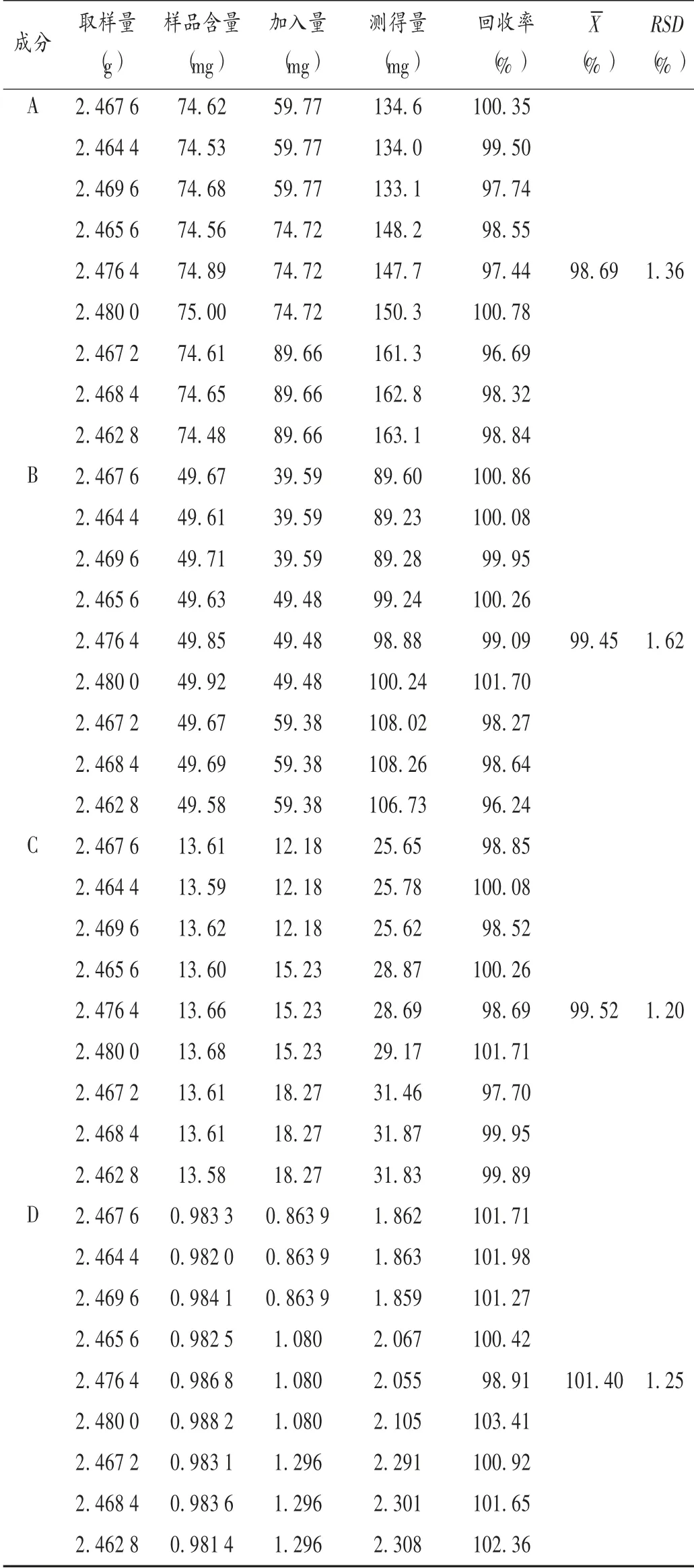
表1 加样回收试验结果(n=9)

表2 2种方法样品含量测定结果(%)
3 讨论
3.1 薄层色谱法建立
酚氨咖敏胶囊现行标准[4]采用薄层色谱法鉴别4种成分,展开剂为三氯甲烷-甲醇-丙酮-乙醚-氨水(9∶0.8∶1∶3∶0.012),该方法要求展距达到15 cm,耗时较长,且展开后马来酸氯苯那敏的Rf值较小。经反复试验和筛查,最终确定三氯甲烷-甲醇-丙酮-乙醚-氨水(9∶0.8∶1∶5∶0.2)为展开剂。当展距达到8 cm时4种待测成分即可达到有效分离,操作简便快速,适宜于本品的快速鉴别。
3.2 含量测定方法选择
在酚氨咖敏颗粒现行标准的色谱条件下,咖啡因和对乙酰氨基酚色谱峰不能达到有效分离,无法准确定量[5]。本研究中建立的气相色谱外标法,可实现1次制备样品、1次进样同时测定4种组分,且10 min之内即可达到有效分离,操作更加简便、快速,亦可用于咖啡因及马来酸氯苯那敏含量均匀度的测定[6]。
3.3 提取溶剂选择
本研究结果比较发现,氨基比林和咖啡因的测定结果接近,马来酸氯苯那敏的测定结果差距较大。现行标准马来酸氯苯那敏的含量测定选用乙腈为溶剂提取后测定。张轶华等[7]报道,用乙腈作溶剂提取马来酸氯苯那敏,提取率较低。本研究中对标准方法提取剩余残渣中马来酸氯苯那敏进行考察,测定剩余量约相当于标示量的6%,表明此方法提取不完全,致使其含量测定结果偏低。马来酸氯苯那敏在乙醇中易溶[8],超声提取20 min即可提取完全。
猜你喜欢
中国药业(2022年22期)2022-12-08
保健与生活(2022年12期)2022-06-09
四川大学学报(自然科学版)(2022年1期)2022-02-10
化学工程师(2021年9期)2021-10-14
蚌埠医学院学报(2020年11期)2020-12-17
分析仪器(2020年5期)2020-11-09
保健与生活(2020年4期)2020-03-02
土壤(2019年6期)2020-01-06
家庭科学·新健康(2018年11期)2018-11-16
发明与创新·中学生(2017年1期)2017-01-20
