
优化的近红外光谱LS-SVM模型测定小麦蛋白质
2020-01-13 01:46陈素彬胡振
食品工业 2019年12期
陈素彬,胡振
南充职业技术学院(南充 637131)
小麦是人类的主粮之一,也是重要的动物饲料、食品原料和外贸商品。蛋白质含量是小麦的基本品质指标之一,在很大程度上反映了小麦的营养价值和加工特性,对其品种选育、栽培管理、市场交易和食品加工等相关环节都有重要意义。
小麦蛋白质含量测定的最新国标方法为凯氏定氮法、分光光度法和燃烧法[1],它们皆为湿化学分析方法,测量精度高,可用于仲裁检验,但有操作复杂、耗时长、污染环境等缺点[2],不适用于大量样本的快速检测。近红外光谱(Near infrared spectroscopy,NIRS)分析是目前应用广泛的仪器分析技术之一,具有快速、方便、简单、准确以及可同时分析多种成分的优点,是一种非破坏性的“瞬间分析”技术[3],能对各种气、液、固态样品进行定量和定性分析,故其被确定为小麦蛋白质含量快速测定方法的国家标准[4]。
近红外光谱分析技术的应用也存在一些问题,如数据量大、谱峰重叠严重、测定结果受环境影响大等[5]。因此,为了在小麦蛋白质含量及其近红外光谱之间建立一个准确高效、泛化性能良好的校正模型,应选用先进的建模方法,并辅之以数据预处理、特征波长选取和模型参数优化等技术手段。已有的一些相关研究采用偏最小二乘(Partial least squares,PLS)[6-7]或人工神经网络(Artificial neural network,ANN)[8-9]方法建模,以无信息变量消除(Uninformative variables elimination,UVE)或连续投影算法(Successive projections algorithm,SPA)筛选特征波长[10-11],获得了较好的结果。但当物质的待测属性与光谱数据之间的线性相关度较差时,不适合建立线性回归模型[12];而ANN模型则存在训练效率低、易发生过拟合现象等缺陷,且非线性模型的参数优化则一直是个难题,常用的几种方法都有明显缺陷,不易确定最优参数值[13]。UVE基于对PLS回归系数b的分析,因此可能所得波长变量仍然较多;SPA则在剔除共线性波段的同时会损失一些有用的光谱信息,且保留的波段中既有待测成分信息,又有其他属性信息,这些都在一定程度上影响了所建校正模型的精度和效率。
试验在通过对比确定小麦样品光谱预处理和样本集划分的最优方案之后,尝试以最小二乘支持向量机(Least squares support vector machine,LS-SVM)建模,并利用改进的二进制蝙蝠算法(Improved binary bat algorithm,IBBA)进行特征波长和模型参数的联合优化,得到一个快速、稳健的小麦蛋白质近红外光谱定量校正模型。所用软件工具主要为MATLAB R2015b,LS-SVMlab Toolbox 1.8,The Unscrambler X 10.4和OriginPro 8.0。
1 材料与方法
1.1 样品采集与制备
收集不同产地、品种的248个成熟小麦样本,剔除杂质后自然晾晒风干,每个样本取1 kg以四分法分成2份:一份用于蛋白质含量的化学方法测定;另一份避光保存于4 ℃冷藏箱中,用于近红外光谱扫描。
1.2 蛋白质含量参考值测定
按照国家标准GB 5009.5—2016《食品安全国家标准 食品中蛋白质的测定》第一法 凯氏定氮法,测定各样品的蛋白质含量。
1.3 近红外光谱采集
每个样品约取500 g,以FOSS公司的Infratec 1241型近红外光谱谷物品质分析仪扫描,波长范围为570~1 100 nm,步长为0.5 nm,扫描10次,取其平均值为样品光谱。248个样品的1 061个波点光谱如图1所示。
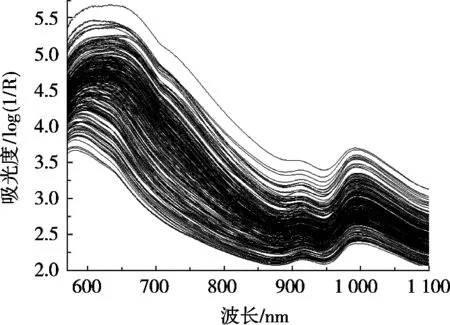
图1 样品原始光谱
1.4 光谱预处理与样本集划分
首先用均值中心化(Mean centering,MC)算法对样品光谱进行预处理,以消除多重共线性的影响。然后以MC预处理结果为基础数据,分别尝试用SG平滑(Savitzky-Golay smoothing,2阶、5点)、标准正态变量变换(Standard normal variable,SNV)、去趋势(De-trending,2阶)、多元散射校正(Multiplicative scatter correction,MSC)和正交信号校正(Orthogonal signal correction,OSC)算法进行处理;分别以CG(Concentration gradient)法、RS(Random sampling)法、KS(Kennard-Stone)法和SPXY(Sample set partitioning based on joint x-y distance)法,按3︰1划分样本校正集和测试集,然后用预处理后的数据建立PLS模型,通过结果比较确定最优的光谱预处理方案和样本集划分方法。
1.5 近红外光谱校正模型建立
LS-SVM能够支持线性和非线性建模,其训练即为求解线性方程组,预测则是计算各建模样本与待测样本之间的核函数,计算量仅取决于校正集的样本数目,而与光谱维数无关。LS-SVM模型比线性模型更稳健,而且避免了ANN模型普遍存在的训练速度慢、易早熟、过拟合和泛化性差等缺陷。但LS-SVM模型(径向基核函数)的核宽度σ2、正则化参数γ与模型的支持向量数目、预测精度、泛化性能密切相关,因此需要寻找这两个参数的最优值;此外,小麦样品的原始光谱中含有大量共线性变量和非目标信息,也会严重影响模型的性能和效率,故应选取与蛋白质含量密切相关的特征波长光谱建模,以摒弃无关信息、降低数据维数,从而在简化模型的同时,提高其精度、速度和稳健性。
1.5.1 改进的二进制蝙蝠算法
蝙蝠算法(Bat algorithm,BA)[14]是Yang基于蝙蝠的回声定位特性提出的一种群体智能搜索算法,其求解准确性和有效性优于常用经典算法,且模型简单、效率高,具有潜在并行性和分布式特性。但是,BA的优化能力主要源于个体间的相互作用,而没有提供能够保持种群多样性的变异机制,故易发生早熟收敛而影响寻优精度,并因强化局部搜索而导致了后期进化变慢[15]。为此,可在BA中引入动态速度权重因子和Cauchy分布随机数扰动,以保持种群的多样性,增强其全局搜索能力,同时加快收敛速度。该算法的原理及执行流程请参考文献[15]。
对改进蝙蝠算法进行离散化,将其搜索空间由连续的实数空间映射到离散的二进制空间,即为IBBA[16]。其主要思想是:蝙蝠的各维位置都限定为二进制值0或1,但其速度则不限制,于是可用传递函数将速度转换为概率值来确定蝙蝠个体的位置。
1.5.2 用IBBA进行建模参数与特征波长联合优化
大多数研究都将LS-SVM的建模参数优化与特征波长选择分别用不同方法实现,但二者在同一个NIRS定量校正模型中是相互影响的[17],将其置于同一过程进行联合优化更易得到最优结果。对此,运用IBBA实现:
以蝙蝠的各维位置对应波长变量,某维取值为1表示相应波长的光谱被选中,反之表示未选中[18]。取值1的各维即为特征波长,将其与建模参数σ2、γ一起构成优化变量,对其进行二进制编码,通过迭代搜索获得其最优值。在每次迭代过程中调用LS-SVMlab Toolbox函数,用校正集样本数据建立LS-SVM模型,将留一法交叉验证(Leave-One-Out Cross Validation,LOO-CV)所得交叉验证均方根误差(Root Mean Square Error of Cross Validation,RMSECV),作为IBBA算法的适应度函数,迭代搜索最优建模参数和特征波长。
1.5.3 优化LS-SVM校正模型的建立与验证
对于经过预处理的校正集样本,取其特征波长光谱数据,以最优参数建立LS-SVM模型,在同等条件下代入测试集样本数据进行验证,并与常用的PLS、CARS-PLS和未优化的LS-SVM建模结果比较。
以校正决定系数Rc2、预测决定系数Rp2、校正均方根误差(Root mean square error of calibration,RMSEC)、预测均方根误差(Root mean square error of prediction,RMSEP)、相对分析误差(Ratio of performance to standard deviate,RPD)为模型评价指标。Rc2和Rp2值大且相差少、RMSEC和RMSEP值小且相差少、RPD值越大,则相应的模型性能越好。
2 结果与分析
2.1 光谱预处理与样本集划分
依照1.4小节的方案,首先分别将原始光谱和MC预处理后的数据以CG、RS、KS和SPXY法划分样本集并建立PLS模型,比较结果得Rp2和RMSEP,发现用MC预处理数据所建PLS模型远优于原始光谱,SPXY法划分的样本集建模效果优于其他方法;然后用相同方法对比“MC+SG平滑”“MC+SNV”“MC+De-trending”“MC+MSC”“MC+OSC”预处理方案,结果以“MC+De-trending”算法最佳,相应PLS模型的Rp2为0.934 0,RMSEP为0.394 0,明显优于其它方案。各种预处理方案的PLS建模结果如表1所示。
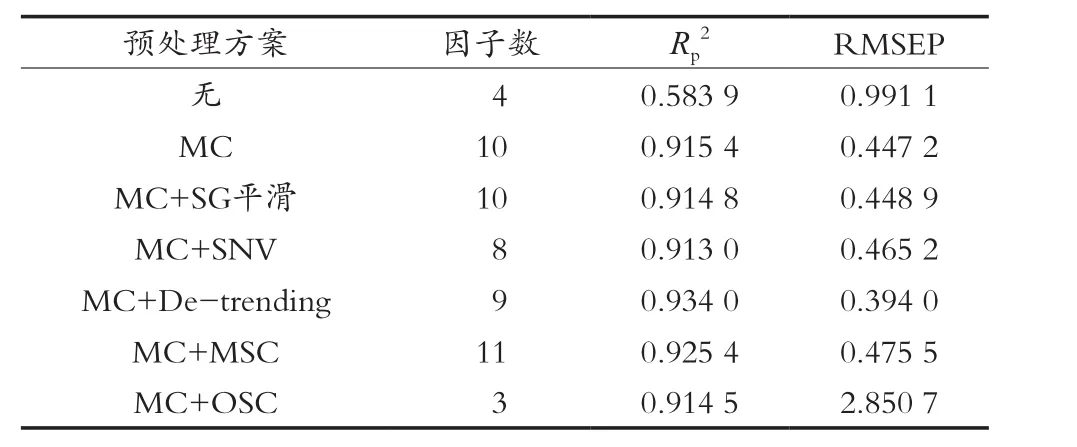
表1 各种预处理方案的PLS建模结果
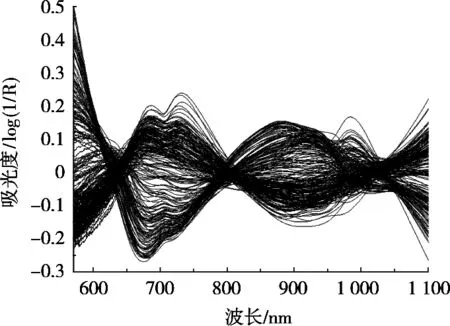
图2 “MC+De-trending”预处理后的谱图
以SPXY法划分的样本集基本信息如表2所示。蛋白质含量最低和最高的样本都被划分到校正集,测试集样本的蛋白质含量在校正集样本蛋白质含量区间之内,表明两个样本集的待测属性值分布合理,所建校正模型能产生较好的预测结果。
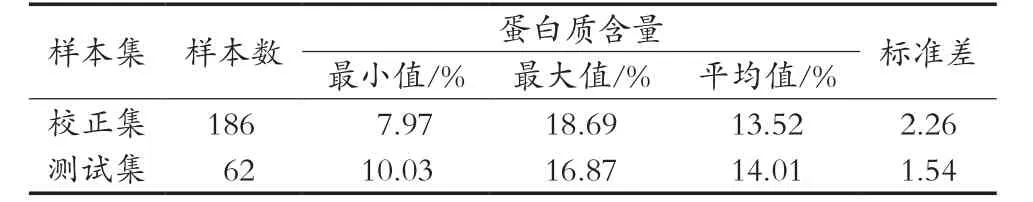
表2 SPXY法划分的样本集基本信息
2.2 建模参数与特征波长优化
按1.5.2小节的方案设计优化流程,用MATLAB编程实现。算法的运行参数设置:迭代次数60,蝙蝠数量40;频率Q∈[0,2],响度初值A=0.25,衰减系数α∈[0.001, 0.999],脉冲频率初值r0=0.1,增强系数β∈[0.001, 1],权重因子w∈[0.5, 1]。
将优化程序运行100次,获得最小适应度函数值时的模型参数值即为最优,其结果为:γ=1 098 472.606 317,σ2=216.010 717。同时选取的建模特征波长为487个。相应寻优迭代过程中RMSECV值的变化如图3所示。
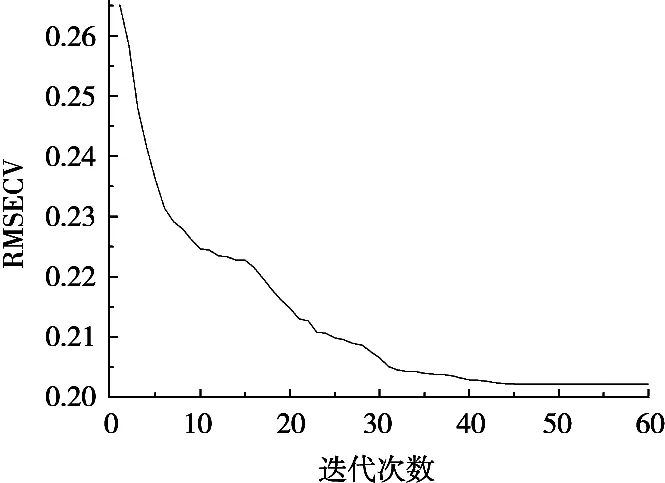
图3 寻优迭代过程的RMSECV值变化
同样地,亦可直接采用适应度函数值最小时的特征波长作为最终建模的波长变量。但IBBA作为一种智能搜索算法,其结果并非唯一确定的,故将100次优化运算产生的100组特征波长进行统计,得到各波点光谱被选中建模的次数,如图4所示。
从图4可以看出,在短波近红外谱区建立小麦蛋白质近红外校正模型时,用IBBA算法选中建模的高概率波长点较分散,相对集中的波段主要有595~599,897~909,916~929,977~1 008和1 012~1 023 nm,此外在576.5,595,598,598.5,600,605,623,625,646,654.5,659和674.5 nm处的光谱被选取的概率也在90%以上。
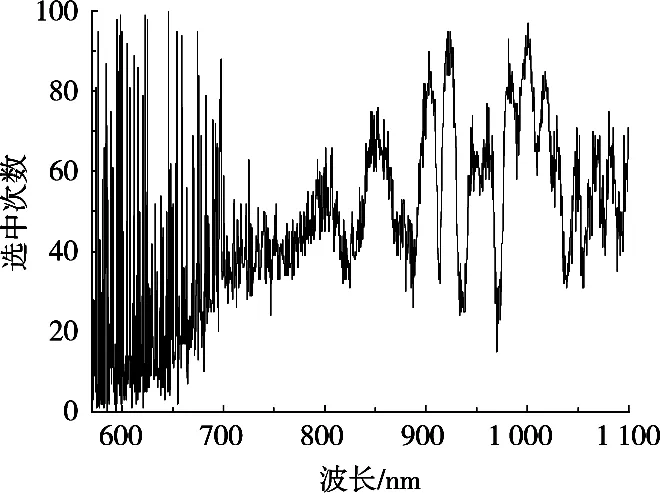
图4 各波点被选中建模的次数
2.3 优化模型建立与验证
以优化所得σ2、γ参数值和特征波长光谱对校正集数据建立LS-SVM模型,再用测试集数据验证其性能,所得结果随波长变量的选取方法不同而有所差异。
方案A:直接采用优化所得487个特征波点建模、验证,其结果为:=0.994 5、RMSEC=0.153 2,=0.983 4、RMSEP=0.197 6,RPD=7.81。
方案B:根据100组特征波长的统计情况选取波长变量。将被选概率50%的波点作为起点,分别向递增、递减两个方向调整特征波长数,用相应光谱数据建模并验证,结果以被选概率为47%以上的对应波点建模最佳:Rc2=0.995 8、RMSEC=0.145 3,Rp2=0.984 2、RMSEP=0.192 4,RPD=8.02。选取不同波长变量所建模型的性能对比如表3所示。
表3表明,当选取的特征波长被选概率从43%开始依次递增时,所建模型的Rp2逐渐增大,且与Rc2的差值逐渐缩小;RMSEP逐渐减小,且与RMSEC的差值逐渐缩小,同时RPD也随之递增,反映出模型的预测精度越来越高,稳健性逐渐增强;但达到47%之后,如果继续提高建模波长的被选概率,则模型的Rc2、Rp2和RPD逐渐减小,RMSEC和RMSEP逐渐增大。究其原因,应是随着选取波点数的持续减少,建模所用光谱数据对于待测属性信息的反映越来越不充分,故而模型性能呈下降趋势。方案B所建最佳模型的小麦蛋白质测试结果如图5所示。该模型对测试集样本预测所得小麦蛋白质含量值位于两条坐标轴的对角线附近,并均匀分布在拟合线两边,表明其精确度高、稳健性好。
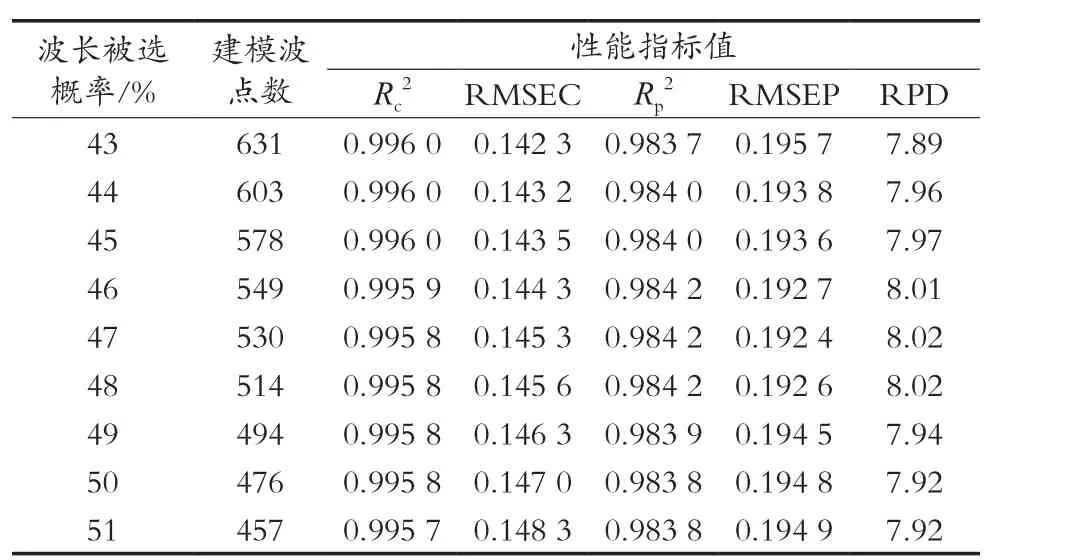
表3 方案B不同波长变量所建模型的性能对比
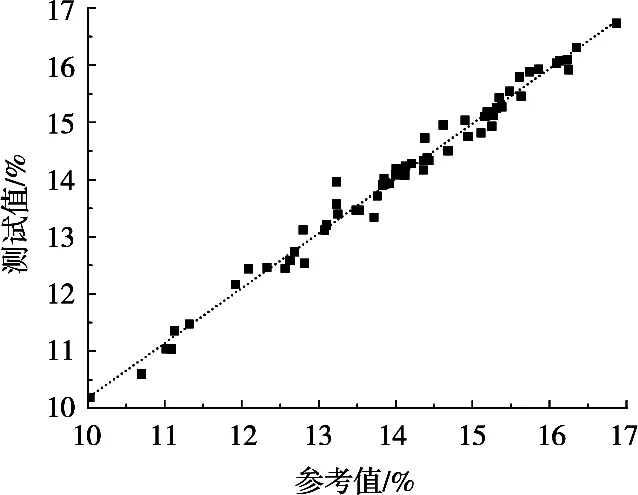
图5 方案B所建最佳模型的测试结果
2.4 与其他建模方案的比较
为进一步确定优化LS-SVM模型的效果,还分别用CARS-PLS和未优化的SVM(Support Vector Machine,支持向量机)、LS-SVM方法对相同的样本集建模并验证,与优化LS-SVM模型及表1的PLS模型进行性能比较,结果如表4所示。CARS-PLS模型选取的特征波长数最少,各项性能指标明显优于PLS模型,其RPD值达到了ICC(International association for cereal science and technology,国际谷物科技协会)标准等级“好”;未经优化的SVM模型则表现不理想,这也正好印证了参数优化对于建立非线性模型的重要性。相比之下,LS-SVM模型的性能优于前三种模型,充分体现其用于小麦蛋白质近红外光谱分析的方法优势;但未经优化的LS-SVM模型稳健性尚有欠缺,故其预测表现较建模指标有明显下降。采用此次试验建立的方法获得最优建模参数和特征波长之后,LS-SVM模型的稳健性和预测精度显著提高,尤以方案B对应的模型性能最佳,其Rc2和Rp2分别达到0.995 8和0.984 2,RMSEC和RMSEP分别为0.145 3和0.192 4,两对指标值都相差很小,且RPD值为8.02,已达到ICC标准等级“非常好”。
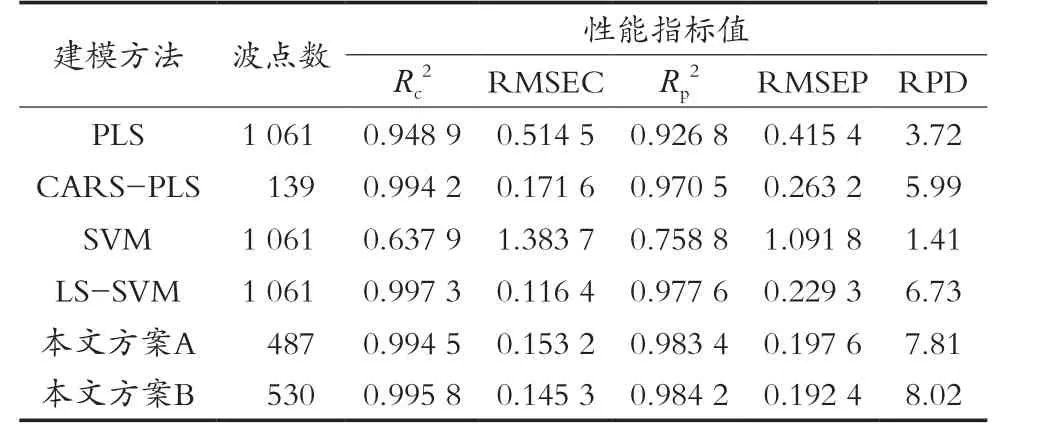
表4 几种方法所建模型性能比较
3 结论
以248个小麦样品为试验材料,通过反复对比,选定“MC+De-trending”算法进行光谱预处理,SPXY法划分样本集;以建模参数和特征波长为优化变量,应用改进的二进制蝙蝠算法搜索其最优值,用之于LS-SVM模型的建立和验证。结果表明,该优化LS-SVM模型性能优异,可用于实际检测工作。
猜你喜欢
特产研究(2022年6期)2023-01-17
国学(2020年1期)2020-06-29
数学物理学报(2017年6期)2018-01-22
制导与引信(2017年3期)2017-11-02
实用口腔医学杂志(2017年6期)2017-09-19
摄影之友(影像视觉)(2017年1期)2017-07-18
中国照明(2016年4期)2016-05-17
工业设计(2016年11期)2016-04-16
环境科技(2015年6期)2015-11-08
物理实验(2015年9期)2015-02-28
