
ATRA对肝癌细胞HepG2生物学行为的影响
2020-01-01 09:07:24张素梅
安徽医科大学学报 2019年12期
周 青,魏 翔,张素梅,汪 渊
目前世界每年新增加的癌症患者约1 808万,死亡约956万[1],在未来相当长的时间癌症仍将是人类面临的一项严峻挑战。随着现代医学的进步人们对癌症的本质有了深入的了解,肿瘤的治疗也由传统的手术、放疗和化疗发展到多学科联合治疗,并出现了诱导分化治疗。诱导分化治疗是指诱导分化剂作用于肿瘤细胞后肿瘤细胞重新向正常细胞分化,生物学特性逐渐向正常细胞靠拢,甚至转变成正常细胞的现象。维甲酸类化合物是典型的诱导分化剂且在临床广泛使用,它包括全反式维甲酸(all-transretinoic acid,ATRA)、3,4-双脱氢维甲酸(ddRA)、9-顺维甲酸(9-cis-RA)和13-顺维甲酸(13-cis-RA)等[2]。研究[3]表明ATRA对多种肿瘤细胞具有诱导分化、抑制增殖并促进凋亡的作用,本文主要研究ATRA作为体外诱导分化剂对人肝癌细胞株HepG2在分化、凋亡、迁移和克隆形成等细胞生物学行为方面的影响。
1 材料与方法
1.1 材料与试剂人肝癌细胞株HepG2(安徽医科大学分子生物学实验室保存);DMEM培养基(美国Gibco公司);胎牛血清(美国Clark公司);ATRA和MTT试剂(美国Sigma公司);细胞凋亡Hoechst 33258试剂盒、BCA蛋白定量试剂盒(上海碧云天公司);γ-GT、AFP检测试剂盒(上海复兴长征医学科学有限公司)等。
1.2 主要实验仪器二氧化碳培养箱、高速冷冻离心机和酶标仪(美国Thermo公司);超净工作台(苏州净化设备公司);荧光显微镜DM400B和倒置显微镜DMI3000B(德国Leica公司);超声破碎仪(宁波新芝公司)等。
1.3 实验方法
1.3.1细胞的复苏和培养 将保存的HepG2细胞从液氮中取出,37 ℃水浴融化,加入含有10%胎牛血清的DMEM培养基中,混匀。置于37 ℃、5% CO2培养箱中,培养3 d传代后使用。
1.3.2细胞形态的观察和AFP、γ-GT比活性的检测 将HepG2细胞分为对照组(含0.1%的DMSO)和ATRA加药组(ATRA溶解于DMSO中,ATRA终浓度分别为1 μmol/L和10 μmol/L),预实验表明0.1%DMSO对细胞生长没有影响。待细胞密度达到80%~90%时,弃去培养液,用PBS洗涤3遍,胰酶消化,DMEM完全培养基终止消化,计数,用DMEM完全培养基调整细胞密度为6×103个/ml,细胞按2、5、14、42、72 d时间培养,2、5 d加药组直接在24孔板中培养,14、42、72 d加药组先在培养瓶中加药培养,3 d传代1次,最后5 d移至24孔板中培养(调整细胞密度为6×103个/ml)。到达预定培养时间后在倒置显微镜下观察细胞形态学的变化并拍照;收集对照组和加药组细胞,离心取上清液检测AFP含量;将细胞沉淀用PBS洗涤3次,加入含0.5%Triton X-100的Tris-HCl缓冲液,超声破碎,4 ℃高速离心15 min(15 000 r/min),取上清液用BCA法蛋白定量后检测γ-GT比活性。
1.3.3细胞增殖实验 细胞分组、培养方法和培养时间同上;取200 μl细胞密度为5×103个/ml的DMEM完全培养基加入96孔板中,每组6个复孔,在到达培养时间时,每孔加入10 μl MTT(5 g/L),置37 ℃培养箱中继续培养4 h,吸尽孔中培养基, 每孔加入100 μl DMSO,酶标仪检测对照组和加药组吸光度A,细胞抑制率%=(细胞对照组OD570-加药处理组OD570)/细胞对照组OD570×100%,重复3次,计算各组细胞抑制率。
1.3.4细胞凋亡实验 将硅化盖玻片放入清洁液中浸泡,流水冲洗,再用去离子水冲洗,高压灭菌后置于70%乙醇中浸泡,使用时超净台中吹干放入24孔板中,种入细胞;细胞分组、培养方法和培养时间同上,各组细胞在密度达到50%~60%时取出盖玻片放在载玻片上固定并Hoechst染色。荧光显微镜下观察细胞形态(激发波长350 nm,发射波长460 nm),细胞核致密浓染为凋亡细胞。每一组细胞随机拍摄3个视野,细胞计数。重复3次,计算细胞凋亡率,凋亡率=凋亡细胞数/细胞总数×100%。
1.3.5细胞迁移实验 取处于对数生长期的HepG2细胞,胰酶消化,DMEM培养基终止消化,吹打均匀后接种至24孔板中。待细胞全部长满24孔板底部,用200 μl塑料吸头在24孔板每孔中央划出一条直线划痕,洗去脱落细胞,在显微镜下拍照作为0 h,对照组和加药组培养至24、48、72 h时,分别在同一观察点拍照记录细胞划痕处的细胞迁移情况,重复3次。利用Quantity One软件测量各孔多点划痕距离取均值,细胞迁移距离为0 h的距离减去24、48、72 h处理后的距离。
1.3.6细胞软琼脂克隆实验 首先在6孔细胞培养板中铺设0.6%琼脂(每孔3 ml),冷却凝固作为底层琼脂;取处于对数生长期的HepG2细胞,制备成单细胞悬液并稀释到终浓度为1×103个/ml;铺设0.3%上层软琼脂,按1 ∶1比例吸取2×DMEM培养基,加入0.6%软琼脂(70 ℃),迅速混匀再加入细胞悬液(细胞终浓度5×102个/ml),轻轻混匀,按每孔1.0 ml加入6孔板中,6孔板置于湿盒中室温放置30 min,再连同湿盒一起置于二氧化碳培养箱,在到达各培养时间时每孔加入MTT染液1 ml,30 min后观察对照组和加药组细胞克隆的形成状态并拍照。
2 结果
2.1 ATRA对HepG2细胞分化的影响
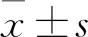
2.1.1HepG2细胞形态学的变化 倒置显微镜下观察到HepG2对照组细胞成簇状生长,细胞密度大,具有良好的光泽度,细胞形态不因培养时间不同而发生变化(图1)。1 μmol/L ATRA组和10 μmol/L ATRA组细胞在第5天时可见细胞数目减少且细胞形态不规则并呈离散状生长,72 d时细胞形态更为细长,多核细胞更少;同一处理时间段10 μmol/L ATRA组细胞形态变化比1 μmol/L ATRA组更加明显。
2.1.2ATRA对HepG2细胞γ-GT比活性和AFP分泌量的影响 与对照组相比,同一处理天数,1 μmol/L ATRA组和10 μmol/L ATRA组细胞γ-GT比活性均呈下降趋势,差异有统计学意义。与对照组相比,1 μmol/L ATRA组和10 μmol/L ATRA组AFP分泌量也呈下降趋势,但处理时间较短时差异无统计学意义;与对照组相比,1 μmol/L ATRA组从第42天起AFP值的差异有统计学意义,10 μmol/L ATRA组从第14天起差异有统计学意义。见表1。
2.2 ATRA对HepG2细胞凋亡的影响在荧光显微镜下可以观察到对照组的HepG2细胞核发出均匀的蓝色荧光(图2),而1 μmol/L ATRA组和10 μmol/L ATRA组可见核致密浓染的凋亡细胞(箭头处为凋亡小体),在第72天10 μmol/L ATRA组凋亡小体最多。统计显示自第5天起1 μmol/L ATRA组和10 μmol/L ATRA组凋亡率高于对照组,差异有统计学意义(表2)。
2.3 ATRA对HepG2细胞增殖的影响MTT实验结果显示,与对照组相比,1 μmol/L ATRA组和10 μmol/L ATRA组从第2天开始生长受到抑制,差异有统计学意义;10 μmol/L ATRA组较1 μmol/L ATRA组对HepG2细胞的抑制程度更高(表3)。
2.4 ATRA对HepG2细胞迁移的影响对照组24 h迁移距离为(7.31±3.20) μm,1 μmol/L ATRA和10 μmol/L ATRA加药组24 h迁移距离分别为(12.28±1.97)μm和(10.50±3.12)μm;48 h对照组迁移距离为(40.03±4.68)μm,1 μmol/L ATRA和10 μmol/L ATRA加药组迁移距离分别为(36.15±4.72)μm和(27.41±2.49)μm; 72 h对照组迁移距离为(50.36±3.14)μm,1 μmol/L ATRA和10 μmol/L ATRA加药组迁移距离分别为(45.76±4.35)μm和(29.63±2.40)μm。统计学分析结果显示,加药组24、48、72 h细胞划痕间伤口愈合距离均低于同期对照组(图3),差异有统计学意义,10 μmol/L ATRA比1 μmol/L ATRA作用更强。
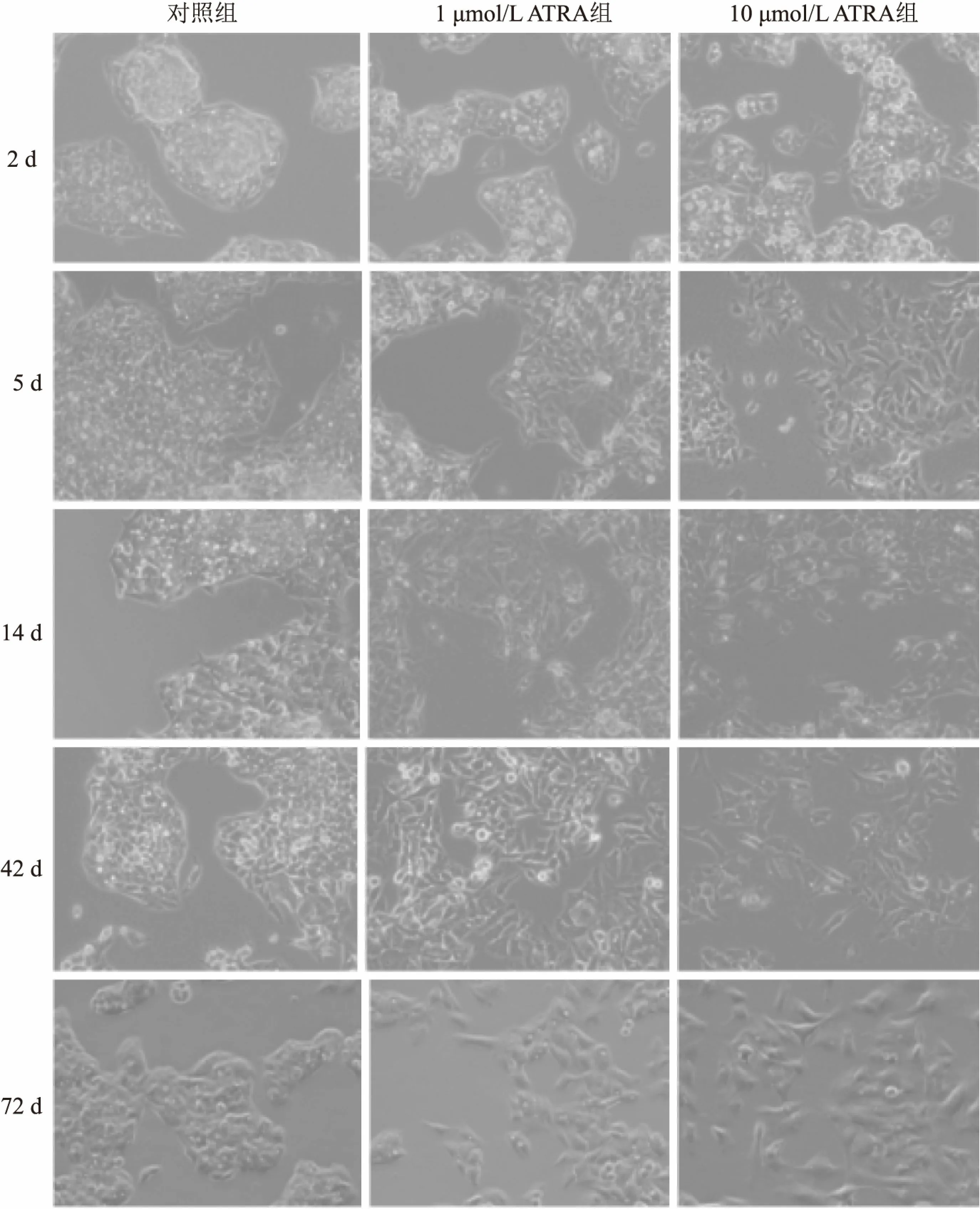
图1 ATRA诱导HepG2分化在细胞形态学上的影响 ×100

表1 ATRA处理肝癌细胞HepG2细胞后 γ-GT的比活性和AFP的分泌量
与对照组比较:*P<0.05
2.5 ATRA对HepG2细胞克隆形成能力的影响软琼脂克隆形成实验表明HepG2细胞可以在软琼脂上形成克隆,与对照组相比,1 μmol/L ATRA组和10 μmol/L ATRA组克隆数目减少,到72 d时1 μmol/L ATRA组仍可见少量克隆,但10 μmol/L ATRA组基本无克隆生长。提示HepG2细胞受到ATRA刺激后能明显抑制HepG2肿瘤细胞在软琼脂中的克隆形成(图4)。

图2 ATRA对肝癌细胞HepG2凋亡的影响 ×200
表2 ATRA对肝癌细胞HepG2凋亡的影响
与对照组比较:*P<0.05
3 讨论
诱导分化剂治疗恶性肿瘤的理论基础是诱导分化剂可以使肿瘤细胞的分化潜能部分或全部恢复进而转化为正常细胞,或诱导肿瘤细胞凋亡从而达到治疗肿瘤的目的[4]。ATRA目前在临床上常被用于急性早幼粒白血病的诱导分化治疗[5]。本研究初步探讨ATRA作用于人肝癌细胞株HepG2后,HepG2细胞分化、凋亡、迁移和克隆形成等生物学行为的变化。
本研究选择γ-GT和AFP作为ATRA诱导HepG2细胞分化的观察指标。正常血清中γ-GT主要来自肝脏,当肝细胞发生癌变时活性逐渐上升且与恶变程度正相关,因此可以把γ-GT作为肝癌诊断的标志物[6]。AFP主要由胎儿肝细胞及卵黄囊合成,正常成人无分泌,且分泌量与肝癌的恶性程度密切相关,AFP是肝癌特异性标志物[7],本研究选择这两个指标作为观察HepG2细胞分化的指标。
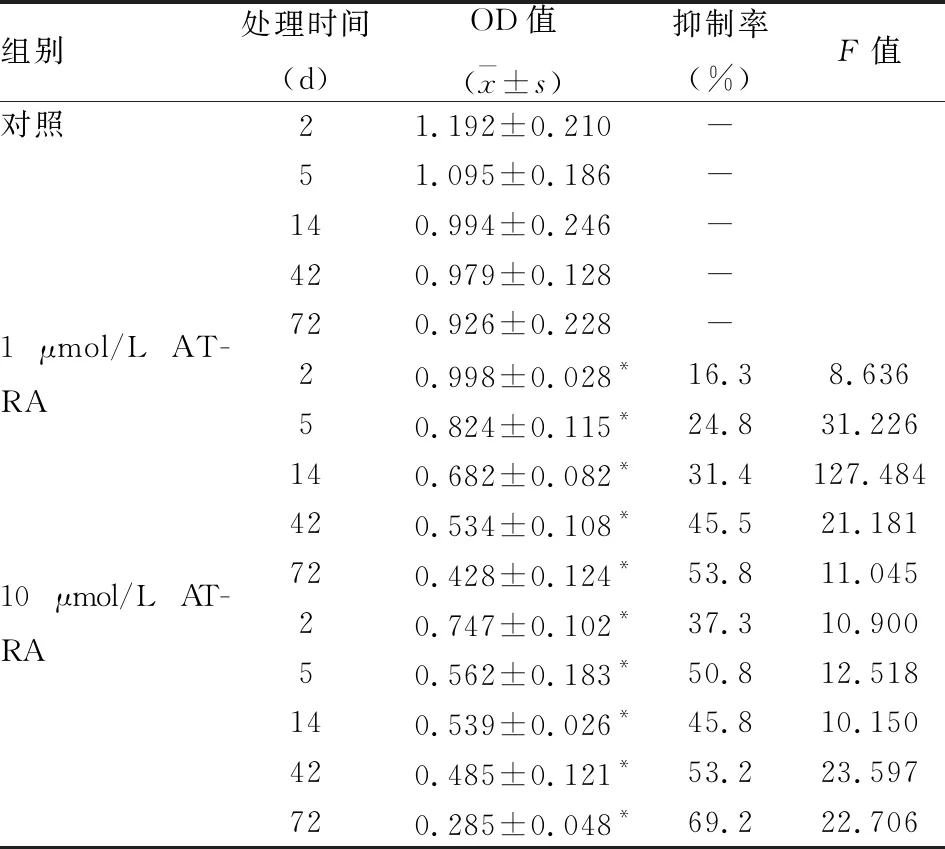
表3 ATRA对肝癌细胞HepG2增殖的影响(n=6)
与对照组比较:*P<0.05
首先观察了1 μmol/L ATRA和10 μmol/L ATRA药物处理组对HepG2细胞分化的影响,结果显示对照组HepG2成簇状生长,细胞密度大,具有良好的光泽度,细胞形态不随培养时间长短发生改变,经过ATRA处理后HepG2细胞生长离散,细胞数目减少,细胞核变小且发生凹陷和分叶,胞质增多,并随着作用时间的延长形态学改变越明显,与Arisi et al[8]的研究中细胞形态学改变一致。结果分析显示:同一处理时间段1 μmol/L ATRA组和10 μmol/L ATRA组的γ-GT比活性和AFP分泌量均低于对照组;组内不同处理时间段之间γ-GT比活性和AFP分泌量也有差异。ATRA加药组在诱导HepG2细胞分化过程中随处理时间的增加γ-GT比活性和AFP分泌量呈下低趋势,表明ATRA处理的HepG2细胞恶性程度降低[9]。提示1 μmol/L ATRA和10 μmol/L ATRA具有诱导HepG2分化的作用。
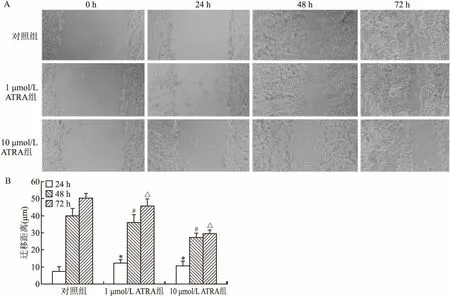
图3 ATRA对肝癌细胞HepG2迁移的影响×100
A:细胞划痕实验图;B:细胞划痕实验柱状统计图;与24 h对照组比较:*P<0.05;与48 h对照组比较:#P<0.05;与 72 h对照组比较:△P<0.05

图4 ATRA对肝癌细胞HepG2克隆形成能力的影响
MTT试验结果表明,ATRA能够抑制HepG2细胞的增殖,与对照组比较,ATRA加药组细胞均自2 d时开始受到抑制,细胞抑制率随处理时间的增加而增加,72 d时细胞抑制率最高达到59.043%。Hoechst染色结果显示细胞对照组在荧光显微镜下细胞核发出均匀的蓝色荧光,偶见凋亡细胞,而加药组可见致密浓染的凋亡细胞、碎裂和凋亡小体。1 μmol/L ATRA处理72 d后,HepG2细胞凋亡率(6.244±1.462)%,10 μmol/L ATRA处理72 d后凋亡率为(9.458±2.246)%。说明ATRA不仅可以促进HepG2细胞向正常细胞再分化、抑制HepG2细胞增殖还可以诱导HepG2细胞凋亡[10]。
根据划痕实验结果可以初步研究肿瘤细胞的迁移能力,通过判断细胞对照组与加药组划痕的愈合距离来判断细胞的迁移能力强弱[11]。实验中观察到在低浓度血清培养基中,1 μmol/L ATRA组和10 μmol/L ATRA组细胞划痕间伤口愈合距离在24、48、72 h均低于对照组;提示ATRA具有抑制HepG2细胞的迁移能力,且随着药物浓度的增加其抑制迁移的作用也随之增强。
HepG2细胞对照组在软琼脂上形成较多的克隆,ATRA刺激后HepG2细胞在软琼脂中的克隆形成减少,72 d后10 μmol/L ATRA组几乎无克隆形成,但1 μmol/L ATRA组仍可见少量克隆。提示ATRA能够抑制HepG2细胞在体外克隆形成能力且ATRA对HepG2克隆能力的抑制程度呈时间与剂量依赖。
猜你喜欢
核科学与工程(2022年3期)2022-10-18 01:25:14
皮肤病与性病(2021年3期)2021-07-30 08:08:48
新农业(2021年9期)2021-06-20 11:26:32
广州化工(2020年6期)2020-04-18 03:30:20
现代矿业(2018年9期)2018-10-16 09:37:02
学生天地·小学低年级版(2017年12期)2018-04-16 03:24:06
食品与生物技术学报(2017年2期)2017-04-09 11:43:29
生物学教学(2016年9期)2016-08-21 02:37:00
初中生学习·高(2016年10期)2016-05-30 22:58:34
物理实验(2015年10期)2015-02-28 17:36:58
