
UPLC-MS/MS 法测定胶基型嚼烟中13 种甜味剂
2019-12-28 03:30:28李国政林奕云蔡群娣张东豫邱建华
烟草科技 2019年12期
付 强,李国政,林奕云,杨 继,蔡群娣,张东豫,邱建华*
1. 广东省测试分析研究所 广东省化学危害应急检测技术重点实验室,广州市越秀区先烈中路100 号34 栋504 510070
2. 河南中烟工业有限责任公司技术中心,郑州经开区第三大街8 号 450000
3. 云南中烟工业有限责任公司技术中心,昆明市五华区红锦路367 号 650031
无烟气烟草制品(Smokeless tobacco products,STPs)是指不经燃烧、直接通过口腔或鼻腔吸食的烟草制品[1]。与传统卷烟相比,无烟气烟草因为具有危害性较小、不产生二手烟、不受场所限制等优点而得到广泛关注[2]。其中,胶基型无烟气烟草制品最早是由瑞典火柴公司首先研发成功并试投放市场[3],是一种以烟草或烟草提取物为有效组分,以可食用胶基为载体,添加了香精香料、软化剂、甜味剂等食品添加剂,通过咀嚼方式向人体递送烟碱的新型烟草制品[4]。近年来,随着全球控烟趋势的加剧及消费者对健康的日益关注,新型烟草制品相关研究已成为烟草行业重要的研究方向之一,胶基型嚼烟分析检测方面的研究目前主要集中在烟碱等成分的分析方面。杨继等[5]用GC-MS 法测定了胶基型嚼烟中烟碱的质量分数;张杰等[6]测定了国外口含型无烟气烟草制品总烟碱、游离烟碱和烟草特有亚硝胺的质量分数;张文娟等[7]研究了胶基型无烟气烟草制品中烟碱的释放行为。
甜味剂是一种重要的食品添加剂,按其来源可分为天然甜味剂和人工合成甜味剂[8],胶基型嚼烟的甜味通常是由添加的甜味剂带来的。与常见的糖类物质相比,甜味剂因具有甜度高、热量低、不易引起龋齿等特点,特别适合在胶基型嚼烟中使用。但是,由于各种天然或合成甜味剂往往带有一定的风味特征或异味,在胶基型嚼烟中添加时,考虑到胶基型嚼烟的风格特征,甜味剂品种和添加量的选择显得尤为重要。美国食品与药品管理局(FDA)对于甜味剂添加限量要求为“适量”添加。我国国家标准对各种食品中甜味剂的添加量进行了明确限定[9]。随着新型烟草制品的研究和开发,急需建立胶基型嚼烟中甜味剂的相关检测指标及方法,为胶基型嚼烟中甜味剂的安全使用提供技术依据。
目前,甜味剂的测定方法主要有离子色谱法[10]、毛细管电泳法[11]、HPLC 法[12-13]、LC-MS 法[14-16]等。由于LC-MS/MS 法的前处理相对简单、选择性强、灵敏度高以及抗基质干扰能力强,目前已应用于食品添加剂的测定[17-19]。但是目前各种方法测定的主要是7 种人工合成甜味剂,尚未见同时测定13 种天然和合成甜味剂的方法报道。本研究中采用UPLC-MS/MS 法对胶基型嚼烟中13 种天然甜味剂和合成甜味剂进行分析研究,旨在为胶基型嚼烟甜味剂的检测提供技术支撑。
1 材料与方法
1.1 材料、试剂和仪器
胶基型嚼烟由云南中烟工业有限责任公司提供,样品先用粉碎机粉碎,选择粒径<5 mm的样品颗粒进行下一步测定。
甲醇、乙腈(色谱纯,美国Honeywell 公司);甲酸(99%)、乙酸(99.8%)(上海晶纯试剂公司);正己烷(AR,广州化学试剂厂);甜蜜素、糖精钠、安赛蜜、三氯蔗糖、阿斯巴甜、山梨糖醇(≥97%,德国Dr.Ehrenstorfer 公司);纽甜(99.0%,加拿大TRC 公司);甜菊糖苷、甘露糖醇、木糖醇(≥98.5%,中国药品生物制品检定所);麦芽糖醇(≥99%,美国Sigma-Aldrich公司);赤藓糖醇、异麦芽酮糖醇[≥99%,美国药典标准品(USP)];水为自制超纯水。
1290 超高效液相色谱仪、6460A 三重四极杆串联质谱仪(美国Agilent 公司);CF16RXII 冷冻离心机(日本Hitachi 公司);Milli-Q超纯水机(美国Millipore公司);BT125D 电子天平(感量0.000 01 g, 德国Sartorius 公司);Oasis HLB 固相萃取柱(美国Waters公司)。
1.2 方法
1.2.1 甜味剂标准储备液的配制
分别称取13种天然和合成甜味剂的标准品,用超纯水配制成浓度均为1.0 mg/mL的标准储备液。分别取木糖醇、山梨糖醇、甘露糖醇、赤藓糖醇、麦芽糖醇、异麦芽酮糖醇储备液,用超纯水稀释配制成浓度均为10 μg/mL的天然甜味剂混合储备液。分别取安塞蜜、甜蜜素、糖精钠、阿斯巴甜、纽甜、三氯蔗糖、甜菊糖苷储备液,用超纯水稀释配制成10 μg/mL 的合成甜味剂混合储备液。所有储备液于4 ℃冷藏保存。
取天然甜味剂和合成甜味剂的混合储备液,用超纯水稀释,配制成浓度分别为10.0、20.0、50.0、100.0、200.0、500.0、1 000.0 μg/L 的混合标准工作溶液。
1.2.2 样品前处理与分析
准确称取1.0 g 胶基型嚼烟样品于50 mL离心管中;加入5 mL 正己烷,涡旋混匀并超声处理10 min;然后加入10 mL 超纯水,振荡提取10 min,涡旋混匀1 min;于3 000 r/min 离心,转移中间水相部分于25 mL容量瓶中。离心管中继续加入10 mL 超纯水重复提取一次,取水相与上一次提取液合并,用超纯水定容。移取1.00 mL提取液于10 mL具塞试管中,加入2 mL正己烷,涡旋混合1 min,静置分层,吸取下层水相过0.22 μm 滤膜,进行UPLC-MS/MS 分析。分析条件:
(1)合成甜味剂。色谱柱:Agilent Poroshell 120 EC-C18柱(4.6 mm×50 mm,2.7 μm);柱温:40 ℃;进样量:10 μL;流动相:甲醇(A),水(B);流速:0.4 mL/min;梯度程序:0~2.5 min 20%~90% A,2.5~3.5 min 90%A,3.5~5.0 min 20%A。
(2)天然甜味剂。色谱柱:GRACE Prevail Carbohydrate ES 柱(4.6 mm×250 mm, 5 μm);柱温:45 ℃;进样量:10 μL;流动相:乙腈(A),水(B);流速:0.4 mL/min;梯度程序:0~5 min 10%B,5~16 min 10%~40%B,16~19 min 40%~75%B,19~20 min 10%B。
质谱离子源:带鞘流气的AJS ESI 源;检测模式:负离子模式;检测方式:MRM多反应监测;干燥气温度:350 ℃;干燥气流量:5.0 mL/min;喷雾气压力:0.31 MPa;鞘流气温度:350 ℃;鞘流气流量:11 L/min;毛细管电压:-3 500 V;喷嘴电压:-500 V。
2 结果与讨论
2.1 甜味剂质谱条件的优化
分别使用浓度为10.0 μg/mL 的天然甜味剂和合成甜味剂混合溶液,通过直接进样的方式在三重四极杆串联质谱仪上对离子源参数、母离子与多反应监测(MRM)模式二级碎片离子的质荷比、碎裂电压(Fragmentor)、碰撞能(CE)等参数进行优化,各种甜味剂的离子信息见表1。本实验中的天然甜味剂和合成甜味剂大多带有羧基或羟基官能团,在电喷雾质谱的负离子模式下可以获得较好的检测灵敏度。

表1 7 种合成甜味剂和6 种天然甜味剂的质谱多反应监测参数Tab.1 MRM parameters of 7 synthetic sweeteners and 6 natural sweeteners
2.2 液相色谱条件的优化
2.2.1 色谱柱的选择
实验中对比了各种不同的色谱柱对合成甜味剂和天然甜味剂的分离效果,合成甜味剂与天然糖醇类甜味剂因分子结构和性质差异较大,只能在不同色谱柱上分别进行分离。分别选用反相色谱柱和糖柱分离合成甜味剂和天然糖醇类甜味剂。
7 种合成甜味剂的分子结构大都具有芳环或杂环结构,且分子结构有明显差异,可以在反相液相色谱柱上实现分离。实验对比了Agilent SB-C18(2.1 mm×50 mm,1.8 μm)、Agilent SB-C18(4.6 mm×100 mm,1.8 μm)、Agilent Eclipse Plus C18ARHD(2.1 mm×50 mm,1.8 μm)、Agilent Poroshell 120 EC-C18(4.6 mm×50 mm,2.7 μm)4 种色谱柱对合成甜味剂的保留和分离能力,结果见表2。综合考虑选用Agilent Poroshell 120 EC-C18(4.6 mm×50 mm, 2.7 μm)柱作为7 种合成甜味剂的分析柱。
天然糖醇类化合物中含有较多的羟基等亲水结构,在常见反相色谱柱和亲水作用色谱(HILIC)柱上均无法进行正常分离。实验对比了两种专用的糖柱Shodex Asahipak NH2P-50 4E(4.6 mm×250 mm, 5 μm)和GRACE Prevail Carbohydrate ES(4.6 mm×250 mm,5 μm)对6 种天然糖醇类甜味剂的色谱分离性能。在Shodex Asahipak NH2P-50 4E 柱上,山梨糖醇+甘露糖醇和麦芽糖醇+异麦芽酮糖醇这两对同分异构体无法实现分离,而GRACE Prevail Carbohydrate ES在乙腈-水流动相体系中可以较好地分离6 种天然糖醇类化合物。 因此选用GRACE Prevail Carbohydrate ES 柱作为天然甜味剂的分离柱。6 种天然甜味剂在不同色谱柱上的分离效果见图1。

表2 4 种反相色谱柱对7 种合成甜味剂的分离效果Tab.2 Separation of 7 synthetic sweeteners by 4 reversed-phase columns
2.2.2 流动相的选择
分别考察了乙腈-水体系和甲醇-水体系作为流动相在Poroshell 120 EC-C18色谱柱上对合成甜味剂的分离效果,分离合成甜味剂的总离子流色谱图和MRM色谱图见图2。结果表明,甲醇-水作为流动相对合成甜味剂的分离度更高,这可以解释为甲醇在反相体系中相对于乙腈具有较弱的洗脱能力,具有一定极性的合成甜味剂在甲醇体系中的保留时间更长,从而表现出更明显的差异性。7 种合成甜味剂的出峰时间在1.5~3.0 min 之间,从总离子流色谱图上看,几种合成甜味剂不能实现完全分离,其中糖精钠和安赛蜜的保留时间重合,阿斯巴甜和三氯蔗糖的保留时间接近。但因为这两种化合物的质荷比相差很大,在质谱MRM 检测模式下基本不存在干扰,保留时间的重合不影响同时测定。
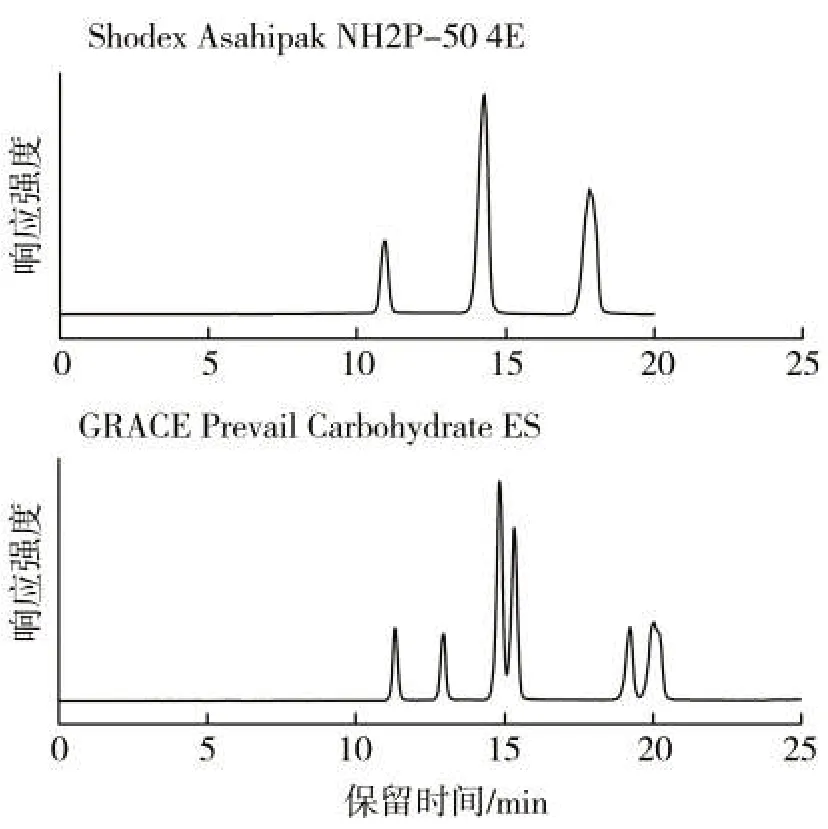
图1 6 种天然甜味剂在不同糖柱上的分离效果Fig.1 Separation of 6 natural sweeteners by different sugar columns
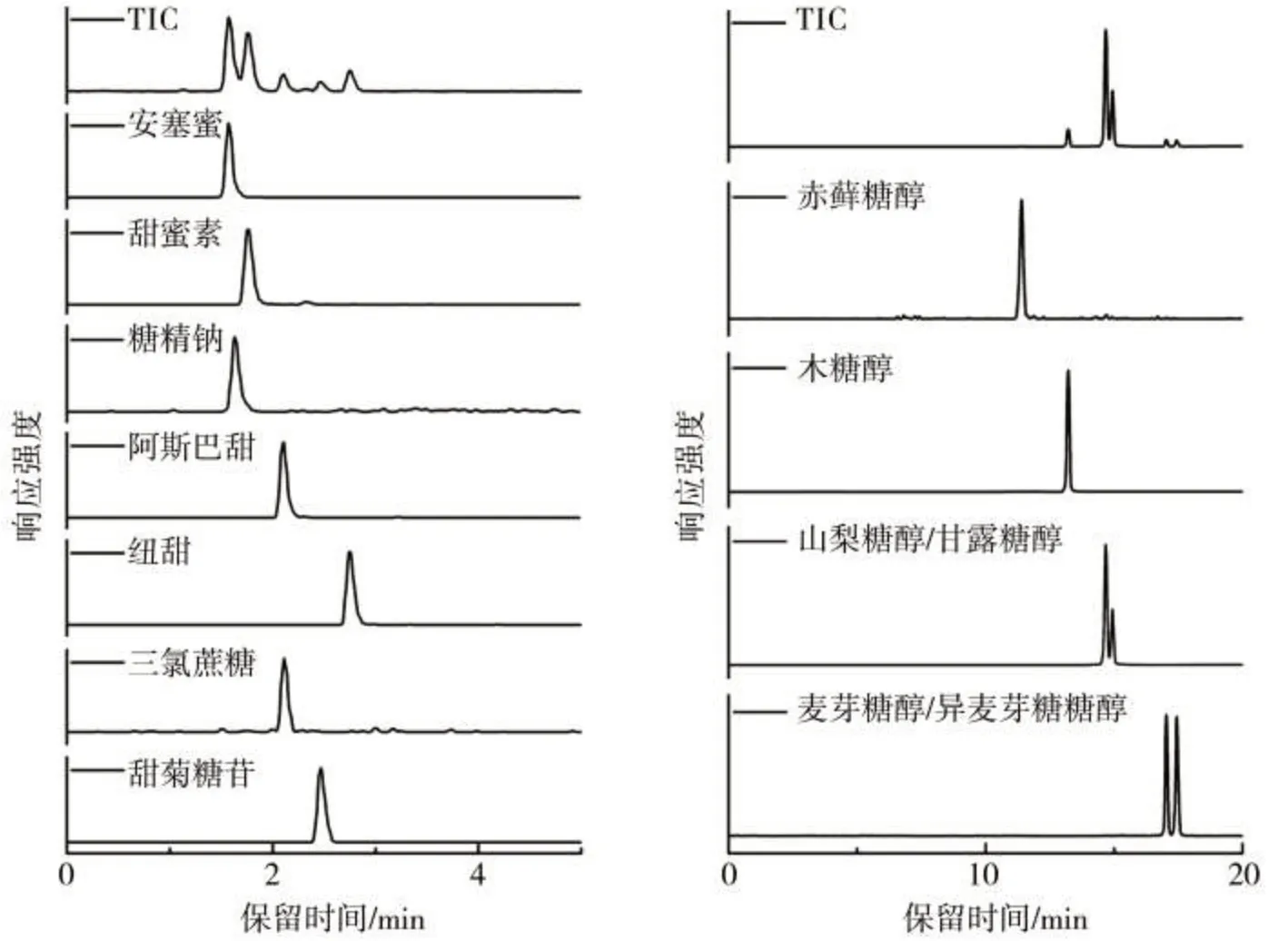
图2 合成甜味剂和天然甜味剂的总离子流色谱图和多反应监测色谱图Fig.2 TIC and MRM chromatograms of synthetic sweeteners and natural sweeteners
另外优化了GRACE Prevail Carbohydrate ES(4.6 mm×250 mm, 5 μm)糖柱在乙腈-水流动相体系下对6种天然糖醇类甜味剂的梯度洗脱条件。天然糖醇的总离子流色谱图和MRM色谱图见图2。在6种天然糖醇类物质中,山梨糖醇和甘露糖醇互为同分异构体,麦芽糖醇和异麦芽酮糖醇互为同分异构体,其结构非常接近且MRM检测参数也几乎一致,无法通过串联质谱相互区分,因此对色谱分离提出了较高的要求。在经过优化的梯度淋洗条件下,6种糖醇类物质的分离效果较为满意,山梨糖醇和甘露糖醇接近实现基线分离,麦芽糖醇和异麦芽酮糖醇完全达到基线分离。
2.3 前处理条件的优化
2.3.1 样品提取条件的优化
本实验中涉及的合成甜味剂和天然甜味剂均易溶于水。测定常规食品中的甜味剂可用水相进行提取;但胶基型嚼烟中的基础物质是胶基,其主要成分是天然树胶、聚醋酸乙烯树脂和丁苯橡胶,还含有橡胶软化剂、增塑剂、乳化剂、填充料等。由于胶基的存在,样品难以被粉碎成细小颗粒且不溶于水,无法直接用水相提取。根据胶基的性质,可以先将样品粉碎成较大的颗粒,然后用有机溶剂溶解、溶胀其中的胶基,再用水相提取。对比了正己烷、四氢呋喃、二氯甲烷对样品中胶基的溶解效果,结果表明,3 种有机溶剂均可快速溶解样品颗粒中的胶基形成悬浊液。综合考虑环境影响和试剂成本,最终选择毒性较小、价格较低的正己烷作为预处理溶剂。
将胶基嚼烟样品用正己烷进行预处理后,可用近似液液萃取的方式对其中的水溶性物质进行提取。对比了使用纯水、甲醇-水(1∶1)、乙腈-水(1∶1)作为提取溶剂对空白加标样品中甜味剂的提取效果。图3 中的结果表明用纯水作为提取剂的提取率高于甲醇/水和乙腈/水,其中原因可能是甲醇/水和乙腈/水体系中溶解了较多的脂溶性成分,在质谱鉴定环节影响了被测物质甜味剂的离子化效率。根据以上结果,采用纯水作为甜味剂的提取溶剂。
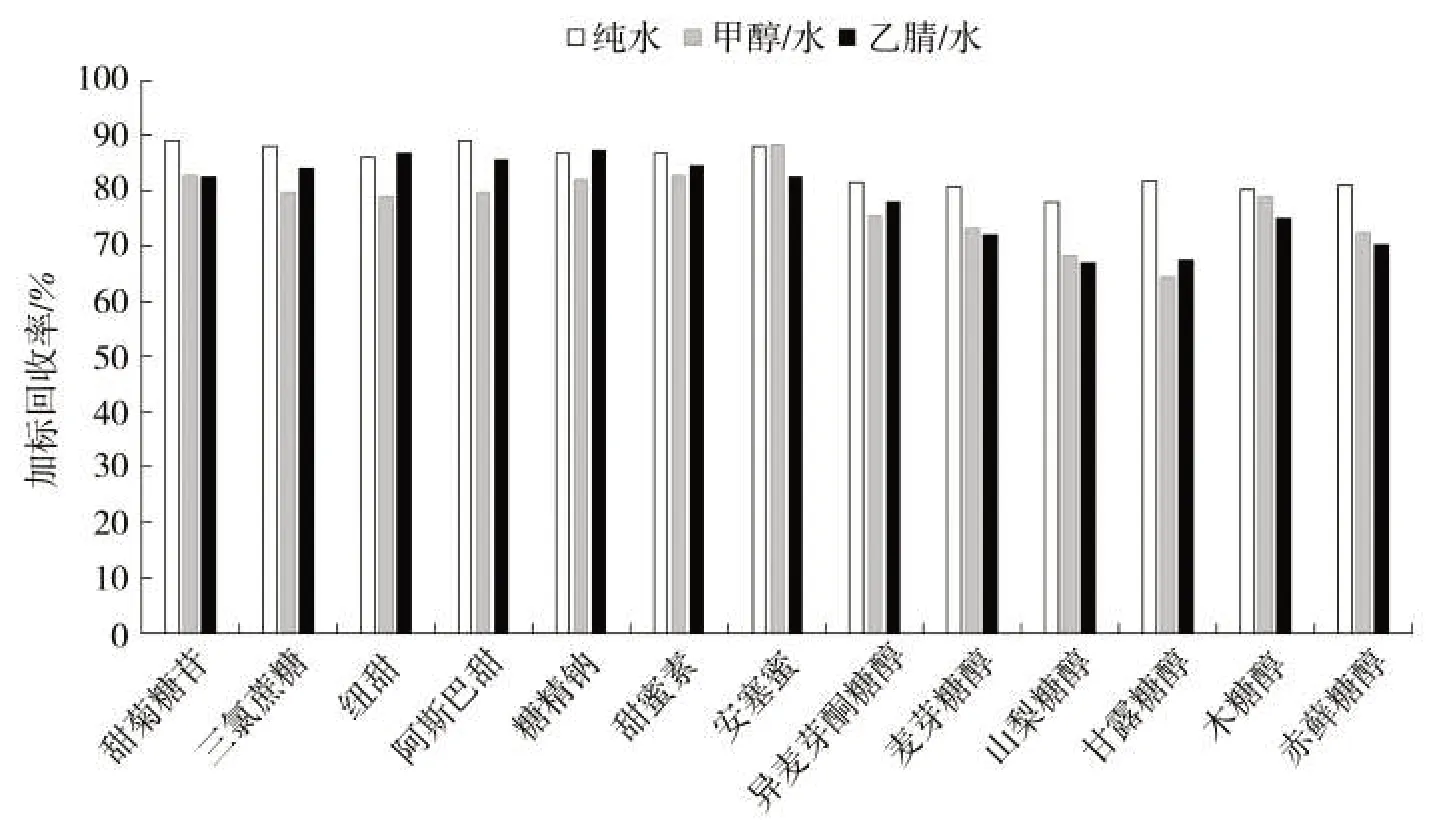
图3 提取溶剂对甜味剂提取效率的影响Fig.3 Effects of extraction solvents on extraction efficiencies of sweeteners
2.3.2 净化条件的优化
胶基型嚼烟类样品中所含的磷脂等脂溶性成分可能在质谱检测时干扰甜味剂的离子化过程,因此有必要对提取液进行净化。实验中对比了不净化直接测定、HLB 柱固相萃取净化[9]和正己烷液-液萃取快速净化对加标样品测定结果的影响,结果见图4。可见,经过Waters Oasis HLB 固相萃取柱净化和正己烷快速净化后的样品提取液,除安赛蜜外的其他甜味剂的响应值均高于未经净化的样品提取液。正己烷净化与固相萃取净化效果相近,且成本低、操作简便,故选择正己烷液-液萃取快速净化方法进行样品提取液的净化。
2.4 线性范围和检测限
按1.2.1 节配制的标准溶液在LC-MS/MS仪上进行分离检测,利用基质加标的方式得出实验的检测结果,然后分别以甜味剂的浓度为横坐标、色谱峰面积为纵坐标绘制标准曲线并进行线性回归。分别按S/N=3 和S/N=10 计算检出限和定量限。各甜味剂的线性方程和相关参数见表3。结果表明,在实验浓度范围内,13 种甜味剂线性回归方程的R2在0.990 7~0.999 6 之间,说明13 种甜味剂标准曲线的线性相关性良好。
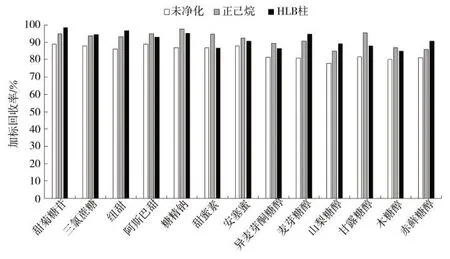
图4 净化条件对13 种甜味剂检测的影响Fig.4 Effects of clean-up conditions on recoveries of 13 sweeteners
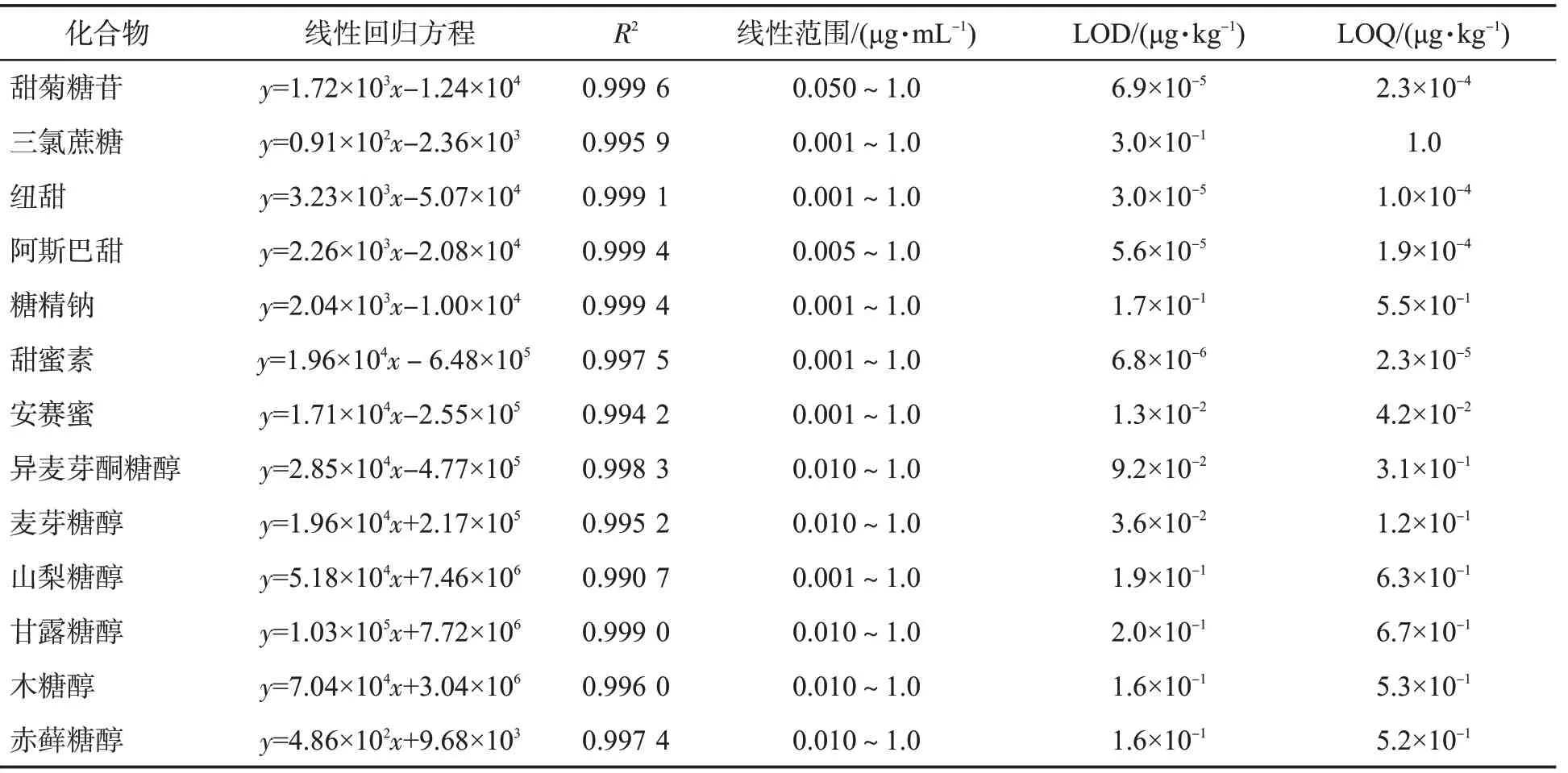
表3 13 种甜味剂的线性回归方程、R2、线性范围、检出限和定量限Tab.3 Linear regression equations, R2, linear ranges, limits of detection, limits of quantitation of 13 sweeteners
经过条件优化下,13 种甜味剂的方法检出限分别在6.8×10-6~0.30 μg/kg之间,定量限分别在2.3×10-5~1.0 μg/kg 之间,优于现行国标方法[9],各甜味剂的线性范围为0.001~1.0 μg/mL,可以满足胶基型嚼烟的检测需求。
2.5 准确性和重现性
经过条件优化,测定5、50、500 μg/kg 3 个添加水平的胶基型嚼烟样品中的各种甜味剂,计算各物质的回收率,结果见表4。可知,在3种添加水平下,13种甜味剂的加标回收率分别在80.2%~95.0%、84.8%~97.6%、83.1%~95.1%之间,说明本检测方法的准确性较高。
为考察方法的精密度,进行了加标样品的日内和日间重复测定,对各样品分别进行6 次平行测定,根据测定结果分别计算各甜味剂的相对标准偏差(RSD)。结果显示,日内、日间RSD范围分别为1.35%~5.56%,3.13%~7.00%,均小于10%,说明本方法重现性较好。
2.6 实际样品测定结果
按优化分析方法测定了10 种胶基型嚼烟样品中各种甜味剂的质量分数,结果见表5。可知,10 种胶基型嚼烟样品中均检出了甜味剂,主要有木糖醇、麦芽糖醇、山梨糖醇、纽甜、糖精钠、安赛蜜。各样品中普遍检出3 种以上甜味剂。其中天然甜味剂的检出现象较为普遍,除甘露糖醇、赤藓糖醇外的其他天然糖醇均有检出,其质量分数在0.9~857 mg/kg范围内。合成甜味剂中三氯蔗糖的检出率相对较高,其质量分数在0.18~1.05 mg/kg 之间。结合嚼烟口味分析可知,具有植物提取物风味的嚼烟中主要添加的是山梨糖醇和麦芽糖醇,而调配的嚼烟中主要是木糖醇和麦芽糖醇。总体而言,检出甜味剂的情况与标签基本相符,且并未超过食品安全国家标准[9]对食品相关甜味剂允许使用量的限值,说明样品中甜味剂的添加情况处于安全范围。
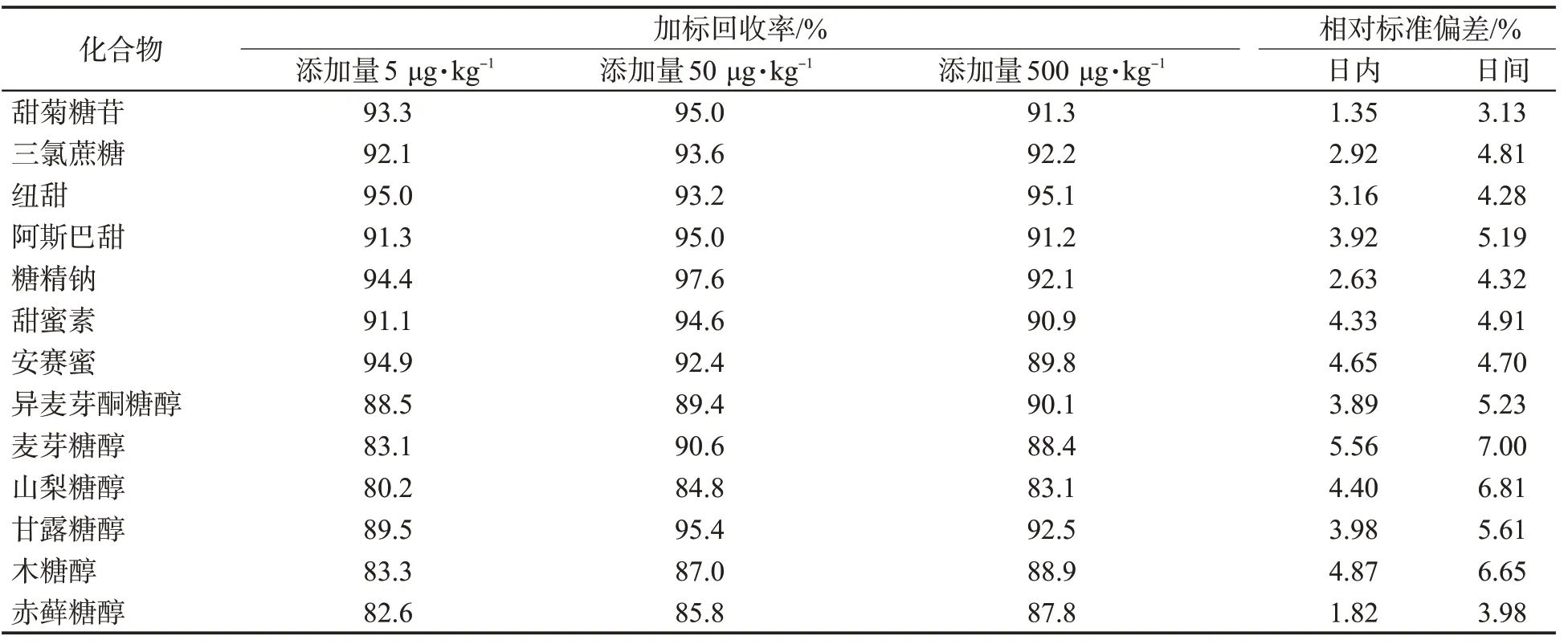
表4 LC-MS/MS 法测定胶基型嚼烟中甜味剂的加标回收率和精密度Tab.4 Spiked recoveries, intra-and inter-day RSDs of sweeteners
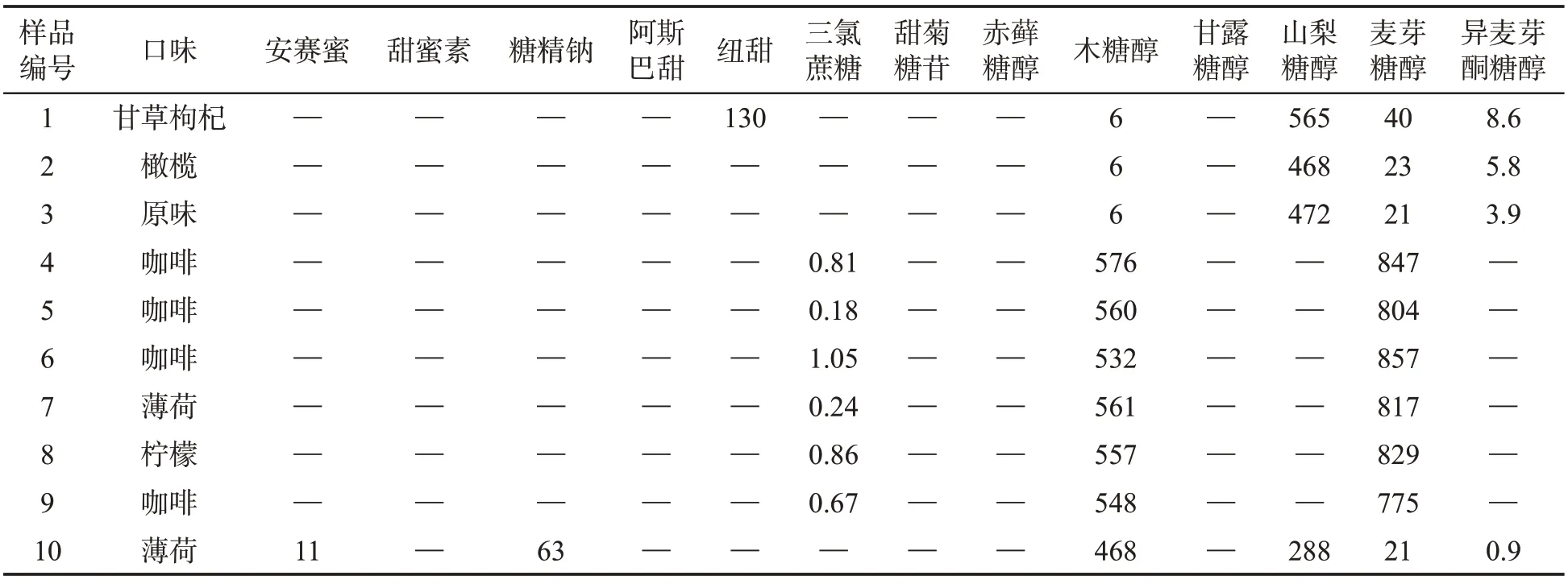
表5 胶基型嚼烟样品中甜味剂质量分数的测定结果Tab.5 Contents of synthetic sweeteners and natural sweeteners in gum-based chewing tobacco samples (mg·kg-1)
3 结论
①优化并建立了测定胶基型嚼烟中7 种合成甜味剂和6 种天然糖醇类甜味剂的UPLC-MS/MS 法:改进了样品的前处理,既降低了基质干扰,又减少了时间和溶剂消耗;优化了反相柱和专用糖柱色谱分离条件,实现了甜味剂组分的完全、快速分离;能满足胶基型嚼烟中甜味剂简单、快速、准确检测的要求;②对10 种胶基型嚼烟中各种甜味剂的检测结果表明样品中甜味剂的添加情况处于安全范围。
猜你喜欢
求学·理科版(2023年6期)2023-04-12 18:35:48
石油炼制与化工(2022年2期)2022-02-15 11:42:26
云南化工(2021年6期)2021-12-21 07:31:04
应用化工(2021年4期)2021-05-20 09:43:36
佛山科学技术学院学报(自然科学版)(2020年6期)2020-11-19 05:55:32
化工管理(2020年26期)2020-10-09 10:05:16
山东化工(2019年2期)2019-02-21 09:29:32
少年科学(2015年10期)2015-10-31 04:19:47
江苏调味副食品(2015年1期)2015-02-28 01:56:34
食品与发酵工业(2014年10期)2014-12-16 08:04:44
