
p53对铁死亡的调节作用及潜在应用*
2019-12-26 01:47:02张丽媛李芙蓉任彩霞
中国病理生理杂志 2019年12期
张丽媛, 李芙蓉, 王 超, 任彩霞
(1北京大学第三医院, 北京 100191; 2北京大学第一医院, 北京 100034; 3北京大学基础医学院组织学与胚胎学教研室, 北京 100191)
p53自被发现以来一直是广受关注的抑癌分子。传统研究发现p53能够诱导细胞凋亡和细胞周期阻滞,从而发挥强大的抑肿瘤作用。当细胞受到外源性或内源性应激因素刺激时,p53可介导细胞周期阻滞,使细胞有足够时间修复DNA损伤,从而防止致癌突变的传播;同时,在DNA损伤无法修复时,p53又可诱导细胞凋亡来清除受损或癌变细胞。但是最近实验数据表明它的上述“经典功能”并不是其抑癌作用的关键,而其在调节细胞代谢等方面的功能可能与肿瘤抑制有更重要的关系[1-2]。铁死亡(ferroptosis)是一种新发现的独特的细胞死亡形式,其与坏死、凋亡、胀亡和自噬等其它细胞死亡形式在形态学、生物途径和遗传上均有本质区别[3-5]。现有研究表明,铁死亡与细胞内代谢状态显著相关[4, 6-7],而p53在细胞代谢的调节中发挥着不可替代的作用[1-2, 8],这提示诱导细胞铁死亡或许是p53发挥抑癌作用的重要机制。
铁死亡是一种铁依赖的脂质氢过氧化物(lipid hydroperoxides)致死性累积引起的细胞死亡形式[4],是一种调节性细胞死亡,受到包括谷胱甘肽(glutathione,GSH)和谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)在内的脂质修复系统的严密调控,并且依赖于含多不饱和脂肪酸(polyunsaturated fatty acid,PUFA)的磷脂的生物合成、PUFA-磷脂酰乙醇胺(phosphatidylethanolamine,PE)的选择性氧化等一系列酶促反应[4]。2012年,有学者首次提出铁死亡的概念,他们在利用高通量小分子筛选技术筛选可以诱导RAS突变的小鼠肿瘤细胞死亡的分子时,发现一种小分子erastin能够抑制细胞膜表面的胱氨酸/谷氨酸反向转运体(cystine/glutamate antiporter,system xc-),导致细胞内半胱氨酸(cysteine,Cys)含量不足。Cys是GSH合成原料,而GSH又是细胞内重要的脂质过氧化物酶GPX4发挥活性的必要成分。因此Cys剥夺会导致GSH缺乏和GPX4活性丧失,最终使细胞内脂质过氧化物沉积,引发细胞死亡。由于这种细胞死亡的发生需要铁依赖的活性氧簇(reactive oxygen species,ROS)的产生,并且铁离子螯合剂能抑制erastin诱导的细胞死亡,表明铁离子在诱导细胞死亡过程中发挥了重要作用,因此他们将这种死亡方式命名为铁死亡[3]。
铁死亡在形态、遗传和机制上不同于细胞凋亡、坏死和自噬等传统的调节性细胞死亡(regulated cell death,RCD)模式[3-5]。铁死亡细胞可以表现出特殊的形态变化,主要表现为细胞变小变圆且相互之间分离、线粒体缩小但线粒体膜密度增加、线粒体嵴减少或消失;而没有核固缩、核溶解和吞噬泡等其他RCD方式的特征性表现[3, 9]。同时,抑制凋亡、坏死和自噬的小分子不能逆转细胞铁死亡结局[3-5]。
1 多种生物学途经可影响细胞对铁死亡的敏感性
细胞对铁死亡的易感性与多种生物学途经相关,包括PUFA代谢、氨基酸代谢和铁代谢等。
PUFA是指含有2个或2个以上双键且碳链长度为18~22个碳原子的直链脂肪酸,其在生理状态下负责调节细胞膜的流动性。PUFA是发生脂质过氧化和铁死亡的重要底物,它的丰度和分布会影响脂质过氧化以及铁死亡发生的程度[7]。游离 PUFA是合成脂类信号分子的底物,它们经过酯化反应变成膜磷脂,膜磷脂可以整合到细胞膜上,调节细胞膜的流动性,也可以进一步被氧化成诱导铁死亡的脂质过氧化物[10]。铁死亡的效应阶段是毒性脂质过氧化物累积的直接结果,脂质过氧化物分解成活性衍生物如醛和ROS,这些活性衍生物会破坏细胞中的蛋白质和核酸[11],从而导致细胞死亡。生理状况下,毒性脂质过氧化物会在GPX4的作用下变成无毒的脂质氧化物,但是当毒性脂质过氧化物产生过多或GPX4活性受损,毒性脂质过氧化物大量累积,便会导致细胞死亡[4]。
GPX4负责降解细胞内有潜在毒性的脂质过氧化物。GSH是GPX4发挥活性的关键,而Cys是GSH的合成原料,因此与Cys、GSH和GPX4相关的氨基酸代谢会影响细胞对铁死亡的敏感性。system xc-是一种跨膜氨基酸转运体,由底物特异性亚基溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)和调节性亚基溶质载体家族3成员2(solute carrier family 3 member 2,SLC3A2)组成,可1 ∶1摄取Cys、排出谷氨酸[12],因此可以通过调节细胞内Cys水平影响GSH活性,进而影响细胞对铁死亡的敏感性。一些细胞可以通过转硫作用合成GSH,因此它们对system xc-抑制剂诱导的铁死亡表现出抗性。Hayano等[13]发现敲除半胱氨酰tRNA合成酶(cysteinyl-tRNA synthetase,CARS)基因可以上调转硫作用,介导细胞对system xc-抑制剂诱导的铁死亡的抗性。此外,过高的谷氨酸水平会产生神经毒性,这与细胞外高水平的谷氨酸水平对system xc-的抑制作用有关[6]。
铁代谢也会影响细胞对铁死亡的敏感性。铁的运入、运出、储存和转换都会影响铁死亡敏感性。脂氧合酶(lipoxygenase,LOX)是一种铁依赖的加氧酶,对脂质过氧化很重要,游离二价铁离子可以通过芬顿反应促进过氧化反应,因此细胞内铁代谢会影响细胞内脂质过氧化物水平和ROS水平,进而影响细胞对铁死亡的敏感性[14]。Shah等[15]研究表明,酶依赖的LOX酶促反应和非酶依赖的铁依赖的自由基反应都在铁死亡中发挥作用。
2 p53增强细胞对铁死亡的敏感性
2015年,Jiang等[16]首次将p53与铁死亡联系起来,认为p53可以通过转录依赖的方式抑制SLC7A11,进而诱导细胞铁死亡。他们将小鼠成纤维细胞和人类某些肿瘤细胞(人乳腺癌MCF7和人骨肉瘤U2OS)中p53的第117、161和162三个位点的赖氨酸残基替换成精氨酸残基,构建成乙酰化缺陷的p533KR突变型细胞,这些细胞丧失了诱导细胞周期阻滞和凋亡等经典的p53功能,但是保留了抑制SLC7A11转录的能力。当用铁死亡诱导剂erastin分别处理p533KR突变型和p53缺失型细胞时,观察到p533KR突变型细胞死亡率大于90%,而p53缺失型细胞死亡率不超过10%,这说明p533KR突变型细胞保留诱导铁死亡的能力。而若在p533KR突变型细胞导入过量SLC7A11后再加入erastin,细胞死亡率会降至20%,抑制SLC7A11转录可能是p53诱导铁死亡的方式。此外,他们还对比了p533KR突变型和p53缺失型细胞系的肿瘤生长速度,发现p533KR突变型细胞系保留肿瘤抑制的能力。2016年,Wang等[17]在p533KR突变型的基础上进一步将细胞中p53的第98位点的赖氨酸残基替换为精氨酸,构建了p534KR98模型,p534KR98模型在丧失p53经典功能的同时也丧失了抑制SLC7A11转录的能力,当用erastin分别处理野生型和p533KR突变型细胞时,他们发现p534KR98模型的细胞死亡率大幅降低,肿瘤抑制功能减退。以上实验结果证明,p53可能通过抑制SLC7A11转录诱导铁死亡,从而发挥抑制肿瘤的作用。
除了SLC7A11,还有一些p53的靶基因可以促进铁死亡,比如谷氨酰胺酶2(glutaminase 2,GLS2)、前列腺素内过氧化物合成酶2(prostaglandin endoperoxide synthase 2,PTGS2)、亚精胺/精胺N1-乙酰转移酶1(spermidine/spermineN1-acetyltransferase 1,SAT1)等。
GLS2可催化谷氨酰胺水解为谷氨酸,它通过降低GSH水平和增加ROS水平来下调细胞的抗氧化防御功能,增加细胞对铁死亡的敏感性。Gao等[6]用谷氨酰胺分解代谢抑制剂可抑制erastin诱导的铁死亡,说明 GLS2升高和GSH降低是铁死亡的必要条件。p53在静息和应激状态下均可增加GLS2表达,表明p53可通过激活GLS2进而大量水解GSH,促进铁死亡[6]。Jennis等[18]发现一种p53变体,该变体可以诱导细胞周期阻滞和细胞衰老,但是不能抑制SLC7A11转录,也不能诱导GLS2表达,该p53变体丧失了肿瘤抑制功能,说明p53通过诱导GLS2或抑制SLC7A11转录诱导铁死亡的能力是其肿瘤抑制机制的关键,而其经典功能对肿瘤抑制不重要。
PTGS2是生物体内前列腺素合成起始步骤的关键酶,它通过调节细胞内关键膜磷脂PE的水平来调节细胞对铁死亡的敏感性。Yang等[19]发现将GLS2和erastin等铁死亡诱导剂作用于p53野生型细胞,该细胞会表现出PTGS2基因表达上调并且发生铁死亡,但用GLS2和erastin诱导p53缺失的细胞,PTGS2基因表达水平不变,并且不会发生铁死亡。所以PTGS2表达上升的过程是p53依赖的,并且与铁死亡直接相关,现在PTGS2被用作铁死亡的标志物[19]。
SAT1是p53的直接靶基因,在p53野生型细胞系中沉默SAT1会降低ROS诱导的细胞死亡率,但是在p53缺失型细胞系中沉默SAT1并不会降低死亡率[20]。SAT1会增加花生四烯酸15-LOX的表达,而花生四烯酸15-LOX是一种铁依赖的PUFA氧化酶,可以增加脂质过氧化,因此SAT1会增加细胞内毒性脂质过氧化物水平,进而增强细胞对铁死亡的敏感性。
综上所述,p53单独并不能诱导铁死亡,而是通过调节基因表达来调节细胞对铁死亡的敏感性。
3 p53延缓细胞发生铁死亡
如上文所述,p53可以增强细胞对铁死亡的敏感性,诱导细胞发生铁死亡,从而抑制肿瘤生长。但另一些研究表明,虽然各细胞系对铁死亡的敏感性不同,但是没有任何一个细胞系可以在p53作用下直接发生铁死亡,证明p53在细胞铁死亡信号调节网络中的作用是复杂而矛盾的[20]。在一些情况下,p53可降低细胞对铁死亡敏感性。Xie等[21]发现p53野生型结直肠癌(colorectal cancer,CRC)细胞对铁死亡诱导剂如erastin不敏感,而通过药物或基因敲除的方式抑制p53活性之后,CRC细胞对erastin的敏感性恢复。Tarangelo等[22]认为稳定持续存在的基础水平p53可以延缓细胞铁死亡的发生。p53在静息状态下会与被其泛素连接酶Mdm2(mouse double minute 2)泛素化降解,因此静息状态下p53无法持续存在。nutlin-3是Mdm2的小分子抑制剂,可使p53在静息状态下稳定存在,而用nutlin-3预处理细胞可以延缓细胞铁死亡的发生,这说明稳定持续存在的p53可以延缓细胞铁死亡的发生。
野生型p53延缓细胞铁死亡的的能力与p21有关[22-23]。p53通过转录依赖的方式激活p21,延迟铁死亡的发生。虽然具体的机制尚不明确,但有一种猜想是p21可作用于GSH,导致细胞内GSH增多,GPX4合成增多,从而使细胞内毒性脂质过氧化物减少,降低细胞对铁死亡敏感性。而在Cys剥夺的情况下,GPX4合成不足,此时野生型p53表达增加会导致细胞铁死亡的敏感性增加。这与之前Jiang等[16]的研究并不矛盾,因为p53无论是通过p21直接调节细胞内GSH水平,还是通过抑制SLC7A11转录增加细胞Cys的水平,其本质都是调节细胞内GPX4的活性和毒性脂质过氧化物的水平,从而影响细胞对铁死亡的敏感性。
另一项研究表明p53可通过与二肽基肽酶4(dipeptidyl peptidase-4,DPP4)结合而抑制CRC细胞铁死亡[21]。DPP4是铁死亡和脂代谢的调节分子,它有肽酶活性,可以降解多种生物活性肽,但是它的酶活性对铁死亡不重要。它可以与烟酰胺腺嘌呤二核苷酸磷酸氧化酶1(nicotinamide adenine dinucleotide phosphate oxidase 1,NOX1)结合,介导DPP4依赖的细胞膜和血浆中ROS生成过程,从而导致细胞内脂质过氧化物大量积累和铁死亡。p53野生型的CRC细胞中,DPP4大量存在于细胞核中,不与NOX1结合,因而ROS合成量较少,细胞对铁死亡敏感性低。而p53缺失或突变的CRC细胞中,DPP4细胞核内沉积受限,从而加速移位到血浆和细胞膜中,与NOX1结合,促进DPP4依赖的脂质过氧化过程,导致细胞铁死亡。DPP4的表达与肿瘤干细胞的存在和不良预后相关。p53调节细胞铁死亡敏感性的不同机制总结如图1。
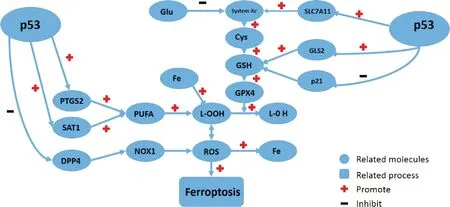
Figure 1.Regulatory mechanism of p53 in ferroptosis. Depending on the condition, p53 promotes or suppresses ferroptosis. Processes positively regulated are shown by “+”, and processes negatively regulated are shown by “-”. PTGS2: prostaglandin endoperoxide synthase 2; GLS2: glutaminase 2; SAT1: spermidine/spermine N1-acetyltransferase 1; PUFA: polyunsaturated fatty acid; SLC7A11: solute carrier family 7 member 11; NOX1: nicotinamide adenine dinucleotide phosphate oxidase 1; ROS: reactive oxygen species; Glu: glutamate; L-OOH: lipid peroxide; L-OH: lipidol; system xc-: cystine/glutamate antiporter; Cys: cysteine; GSH: glutathione; GPX4: glutathione peroxidase 4; DPP4: dipeptidyl peptidase-4.
4 影响p53对铁死亡调节作用的因素
p53对细胞命运的调控是非常复杂而精细的,不同的细胞类型、不同种类的应激因素甚至不同强度的同一应激因素,都有可能激发不同的p53信号通路,引向不同的细胞命运[1]。但从p53庞大繁杂的信号网络中,我们依稀可以识别出各种不同的p53信号通路的共同目的:保持基因组稳定性,抑制肿瘤发生。为了达到这个目的,它会诱导细胞周期阻滞,为DNA修复提供必要时间;会诱导细胞死亡(包括细胞凋亡和铁死亡等),清除基因组异常的细胞;同时,在轻度应激的状况下,它还会促进正常细胞存活。完整的p53信号通路会抑制肿瘤的发生和发展,但p53单个或几个位点的突变可能只影响p53信号网络的一部分,导致原有的p53信号通路不完整。若p53抑制细胞生长增殖的功能丧失,提高细胞存活能力的功能保留,则不仅会加速肿瘤的发生和发展,还会导致耐药[1, 24]。
同理,p53在铁死亡过程中的作用也是如此,它通过几条独立的信号通路分别调控不同细胞类型和不同应激因素下细胞对铁死亡的敏感性,一方面p53可以抑制system xc-,导致Cys剥夺;或作用于GLS2增加GSH水解,导致GXP4失活;或作用于脂质过氧化物合成酶,导致脂质过氧化物合成增加,增加细胞对铁死亡的敏感性。另一方面,当细胞发生Cys剥夺时,另一条信号通路被激活,野生型p53表达增加以诱导p21转录或抑制DPP4与NOX1的结合,最终抑制细胞对铁死亡的敏感性。
这两者看似矛盾,实则统一,一方面p53增加细胞对铁死亡的敏感性,因为作为一种调节性细胞死亡形式,铁死亡在物种进化中有生理意义,p53可以通过增加细胞对铁死亡的敏感性达到其清除异常细胞以及抑制肿瘤发生的目的;另一方面,它抑制铁死亡,因为p53还有一个重要的功能就是帮助基因组正常的细胞度过各种应激因素的伤害。当发生代谢应激时,p53会通过增强细胞对ROS水平的调节能力,降低细胞对铁死亡的敏感性,促进细胞存活。为了适应体内ROS应激和Cys剥夺的体内环境,一些肿瘤会保持p53-p21轴的正常运行,在适当的动物模型中验证这个假说将会是有趣的研究方向。
但是p53对细胞对铁死亡敏感性的调节机制依然不完全清楚。目前认为p53对细胞死亡敏感性的调节可能与以下几种因素有关:(1)细胞类型:不同的细胞类型对铁死亡敏感性不同,p53对铁死亡的作用也不同。虽然p53野生型的CRC细胞对凋亡刺激很敏感,但是它们对铁死亡诱导剂如erastin是不敏感的。而相反,通过药物或基因敲除的方式抑制p53活性之后,CRC细胞对erastin的敏感性恢复,并且同时,这些缺乏p53的CRC细胞对凋亡刺激表现出抗性[21, 25]。在非CRC细胞中,p53增强细胞对铁死亡的敏感性,通过抑制SLC7A11的表达或激活SAT1来促进铁死亡。应该关注动物模型中不同组织的细胞中p53在铁死亡扮演的角色和铁死亡在肿瘤抑制中扮演的角色。这与之前发现的p53在其他非CRC细胞中的作用是不同的,也暗示p53对铁死亡的作用与肿瘤细胞类型相关。(2)p53缺失/突变:正常细胞中,p53增强细胞对铁死亡敏感性和延缓细胞发生铁死亡的信号通路同时存在,并且有着完善的调节机制。因此受到轻中度应激刺激时,p53延缓细胞发生铁死亡的信号通路会被激活,帮助细胞克服应激继续存活。若细胞遭受重度应激,p53会启动增强细胞对铁死亡敏感性的信号通路,诱导细胞发生铁死亡,防止致癌突变的扩散。而在部分肿瘤细胞中, p53突变位点损害了其诱导细胞发生铁死亡的能力,而保留了其延缓细胞发生铁死亡,协助细胞度过应激的能力,这会导致肿瘤细胞过度增殖并且生命力顽强,可能与肿瘤耐药性有关[24]。同时,肿瘤细胞可能在铁死亡其他的信号通路发生突变,直接调节细胞对铁死亡的敏感性。例如它们可表达过量的抗氧化转录因子核因子E2相关因子2(nuclear factor E2-related factor 2,NRF2),在转录水平调节SLC7A11,从而抑制铁死亡。Sun等[26]研究发现,在肝细胞癌中抑制NRF2信号通路,会增加erastin和Sora的抗癌活性。
5 p53对铁死亡调节作用的潜在临床应用
作为一种新发现的细胞调节性死亡方式,铁死亡可发生在神经细胞和血管内皮细胞等正常组织细胞,导致神经退行性疾病(阿尔茨海默病、亨廷顿舞蹈症和帕金森病等)和缺血性疾病(脑卒中、颅内出血、创伤性脑损伤和缺血再灌注损伤)的发生[4, 27]。铁死亡也可能发生在各种肿瘤细胞中,在各种化疗药物耐药导致药物疗效不佳、严重危害病人生命和健康的时候,铁死亡可为诱导定向清除肿瘤细胞和克服肿瘤耐药提供新的研究方向[5, 23, 28]。虽然目前在不同影响因素下p53对铁死亡的调节机制并未完全研究透彻,但这庞大而复杂的信号网络无疑为我们提供了广阔的疾病治疗思路:研究p53对铁死亡调节作用的上游信号通路,了解p53增强或减弱细胞对铁死亡敏感性的影响因素,让p53在“刹车”和“油门”之间灵活转化。针对过度增殖的肿瘤细胞,我们可同时抑制p53延缓铁死亡的信号通路和促进p53激活铁死亡的信号通路,使铁死亡成为继细胞凋亡之后又一重要的化疗药物作用机制。而对于不可再生的神经细胞和心肌细胞,通过对p53信号通路的调节抑制细胞铁死亡无疑是值得尝试的治疗策略。
6 结论
铁死亡是一种独特的RCD形式,其与脂质代谢、氨基酸代谢、铁代谢等多种生物学过程相关。p53是广为关注的抑癌因子,现有研究表明其诱导细胞发生细胞周期阻滞和细胞凋亡等的能力不是其抑制肿瘤生长的关键,而其在细胞代谢调节方面的能力与肿瘤抑制有关。且与以往单一的信号通路不同的是,现有观点认为p53身处一个强大的信号网络的核心,不同的上游信号通路可能会以不同的方式激活p53,从而介导不同的p53下游信号通路。p53可以增强细胞对铁死亡的敏感性,从而阻止基因突变的积累与细胞癌变。同时,p53也可以延缓细胞发生铁死亡,帮助处于应激状态下(如Cys剥夺)的正常组织细胞度过应激状态。目前研究表明细胞类型和p53突变位点可能影响p53对细胞对铁死亡敏感性调节的选择机制,但是更为具体确定的选择机制有待进一步研究。研究清楚p53在因素作用下对铁死亡的调节作用,有助于我们选择特定的信号通路作为靶点,或诱导肿瘤细胞发生铁死亡,作为肿瘤治疗的新靶点;或抑制组织细胞发生铁死亡,以减轻化疗药物的副反应、治疗神经退行性疾病及血管性疾病。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:52
河北科技师范学院学报(2022年2期)2022-08-26 08:55:46
中成药(2018年9期)2018-10-09 07:18:36
中国有色金属学报(2018年2期)2018-03-26 07:58:26
中成药(2018年1期)2018-02-02 07:19:53
中成药(2017年4期)2017-05-17 06:09:26
中国塑料(2016年3期)2016-06-15 20:30:00
焊接(2016年1期)2016-02-27 12:55:37
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:36
新闻传播(2015年8期)2015-07-18 11:08:24
