
外电场作用下1,1-二氯乙烯的结构和光谱
2019-12-26 02:04邱学军聂文星
中南民族大学学报(自然科学版) 2019年4期
邱学军,聂文星
(1中南民族大学 电子信息工程学院, 湖北省智能无线通信重点实验室,武汉 430074;2 四川大学 物理学院,成都 610065 )
1,1-二氯乙烯,又称为偏二氯乙烯,是生产聚偏氯乙烯(PVDC)产品的主要单体,也是生产氯氟烃替代物的一种重要中间体,在医药、染料等行业有着广泛的应用.尽管如此,1,1-二氯乙烯分子是重要的致癌物,2017年,世界卫生组织国际癌症研究机构将其纳入2B类致癌物清单之中.人体在短时间内接触1,1-二氯乙烯,会产生恶心、呕吐甚至酩酊症状,长时间接触则可能产生神经衰弱.此外,在紫外光作用下,1,1-二氯乙烯分子很容易光解产生氯原子[1],这对臭氧层会产生极大的破坏.因此,过去十几年来,1,1-二氯乙烯的光解离动力学受到了人们广泛的理论[2,3]和实验[4-7]关注,如在理论方面,RIEHL 等人利用从头算分子轨道理论研究了1,1-二氯乙烯分子的解离动力学[2],在实验方面,HUA 等人利用离子速度影像和共振增强多光子电离的方法研究了二氯乙烯分子三种不同构型在214.5 nm和235 nm作用的光解离通道[6], MOSS等人利用红外辐射光谱研究了1,1-二氯乙烯分子在193 nm激光作用下的光解离机理等[7].
近年来,人们发现利用外电场作用在分子体系上,可以使分子发生一系列的物理化学变化,如分子键长、能级结构、光谱以及旧化学键的断裂与新化学键的形成等[8-10].研究外电场与分子的相互作用而引起的分子的结构和光谱的变化,对研究外电场作用下分子的光解离和降解等具有十分重要的意义.尽管如此,目前关于1,1-二氯乙烯分子在外电场中的结构和光谱的研究还尚未见文献报道.
本文采用Gaussian 03[11,12]软件对1,1-二氯乙烯分子进行理论计算,得到了分子在不同外电场作用下基态的优化构型、分子键长、总能量、电偶极矩、电荷分布、轨道能级、红外振动模式和红外光谱、分子激发态能量、激发波长和紫外吸收光谱等信息.
1 理论计算方法
分子体系与外电场相互作用的哈密顿量可以表示为[13]:
H=H0+Hint,
(1)
其中H0为无外场作用下的哈密顿量,Hint为分子体系与外场相互作用下的哈密顿量,在偶极近似条件下,相互作用哈密顿量可以表示为[14]:
Hint=-μ·F,
(2)
其中μ表示分子电偶极矩矢量,F表示外场矢量,文中所有外场单位均使用原子单位a.u.,1a.u.=5.1425×1011V/m.
理论计算在Gaussian 03中进行.首先采用DFT方法, 在B3LYP/6-311++G(d,p)高精度基组水平上优化分子结构,得到了不同外加电场条件下1,1-二氯乙烯分子的基态的优化构型、分子键长、总能量、电偶极矩、电荷分布和轨道能级.其次,在优化构型的基础上进行频率计算,得到了分子的红外振动模式和红外光谱,并对主要的特征峰进行了标定.最后,利用TD-DFT方法, 在B3LYP/6-311++G(d,p)高精度基组水平上进行能量计算,得到了不同外加电场下分子的激发态能量、激发波长以及紫外吸收光谱.
2 理论计算结果
2.1 不同外加电场对1,1-二氯乙烯分子的结构和电荷分布的影响
采用DFT方法在B3LYP/6-311++G(d,p)基组水平上计算了1,1-二氯乙烯分子的基态优化构型,如图1所示.
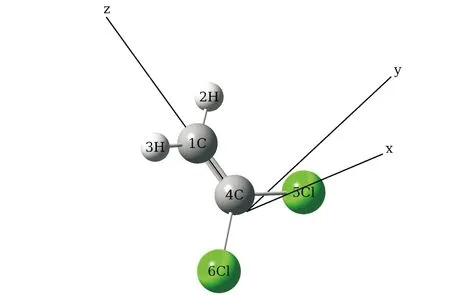
图1 优化的1,1-二氯乙烯分子在无外电场时的分子结构Fig.1 The optimized molecular structures of 1,1-dichloroethylenein the absent of external electric field
在z轴(C=C)方向上加不同强度的外电场(0~0.045 a.u. ),优化获得的结构参数如表1所示.由表1可以看出, 随着电场的增大, C=C键长逐渐增长,C=Cl键长逐渐缩短,C=H键长则略微增加.这些结构的变化主要是由于分子在内外电场的共同作用下的电荷转移而导致的.表2显示了不同外电场对分子电荷分布的影响,由表2可以看出,1C和4C原子均带负电,两个Cl原子和H原子均带正电,随着外电场增大,1C原子电荷逐渐减小,电荷逐渐向4C原子聚拢,且4C原子电荷增加较多,这导致1C和4C原子核排斥力增大,化学键变长.同理,电荷分布变化导致C-Cl原子间原子核吸引力变大,化学键缩短.由于H原子电荷分布基本保持不变,1C原子电荷略微变小,导致C-H原子原子核吸引力减小,化学键略微增加.此外,从表2还可以看出, 随着正向电场增加, 分子总能量略有增高, 这主要是因为电荷分布变化引起C-Cl键长缩短,导致电偶极矩减小, 从而分子的总能量升高.

表1 优化的不同外电场作用下1,1-二氯乙烯的结构参数、总能量和电偶极矩Tab.1 The optimized structure parameters, total energy and dipole moment of 1,1-dichloroethylene in different external electric field
表2 不同外电场作用下1,1-二氯乙烯的电荷分布
Tab.2 The charge distribution of 1,1-dichloroethylene in different external electric field

F/a.u1C4C5Cl6Cl2H3H0-0.797-0.1360.3300.3300.1370.1370.01-0.786-0.1580.3340.3340.1370.1370.02-0.771-0.1800.3390.3390.1360.1360.03-0.759-0.2010.3440.3440.1360.1360.04-0.744-0.2260.3490.3490.1360.1360.045-0.736-0.2390.3510.3510.1370.137
2.2 不同外加电场对1,1-二氯乙烯分子轨道能级影响
分子的最高占据分子轨道(HOMO)和最低未被占据分子轨道(LUMO)能量反映了分子失去或者得到电子能力的强弱,HOMO能量越高,分子越容易失去电子,LUMO能量越低,分子越容易得到电子.分子能隙则反映了分子中电子从占据轨道跃迁到空轨道的概率大小,也可以用来表示分子参与化学反应的能力.采用与2.1中相同的方法,计算获得了1,1-二氯乙烯分子的HOMO和LUMO轨道能量以及能隙的大小,如表3所示,其中能隙通过下式得到:
EG=(EL-EH)×27.2 eV,
(3)
由表3可以看出,随着电场的增大,1,1-二氯乙烯分子的HOMO轨道能量、LUMO轨道能量以及分子能隙均随电场增大而逐渐减小,这意味着在强电场作用下,分子轨道中的电子极容易跃迁到空轨道中,从而导致1,1-二氯乙烯分子被激发而产生解离.
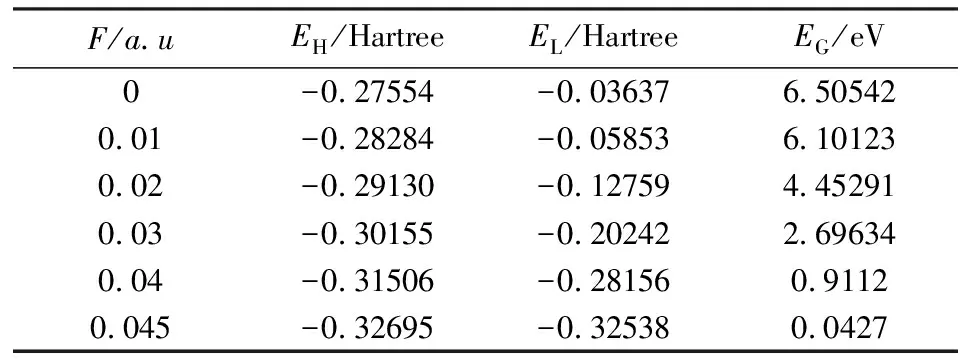
表3 不同外电场作用下的HOMO轨道能量EH、LUMO轨道能量EL以及HOMO-LUMO能隙EGTab.3 The acquired HOMO orbit energy EH, LUMO orbit energy EL, and HOMO-LUMO energy gap EQ in different external electric field
2.3 不同外加电场对1,1-二氯乙烯分子的红外光谱的影响
在分子优化构型的基础上,进一步进行了频率计算,得到1,1-二氯乙烯分子的红外光谱,如图2所示.图2(a)显示了无外电场作用下的红外光谱,其中黑色曲线表示理论计算结果,红色曲线是美国国家标准局(NIST)数据库提供的数据[15],由图可见,理论计算结果与实验结果的谱峰位置吻合得很好,说明我们使用的计算方法和优化的分子基态结构是可信的.
从计算结果来看,1,1-二氯乙烯分子有3个较强的振动特征峰,其对应的波数分别为763 cm-1, 1091 cm-1和1658 cm-1.这三个振动对应的振动方式如图3所示,图中黄色箭头表示跃迁偶极矩的单位矢量,蓝色箭头表示位移矢量.由图可见,763 cm-1和1091 cm-1对应C2H2基团的摇摆振动,1658 cm-1则对应1C=4C双键的伸缩振动和2H、3H原子的摇摆振动.图(b)-(d)分别给出了不同外电场对763 cm-1, 1091 cm-1和1658 cm-1这三个红外振动模式的影响,由图可以发现,外加电场对三个振动模式的影响不尽相同,随着外电场强度的增加,C2H2基团的两个摇摆振动频率逐渐增大,呈现明显的蓝移现象.而C=C双键的伸缩振动模式随电场增大,频率则逐渐减小,呈现明显红移.
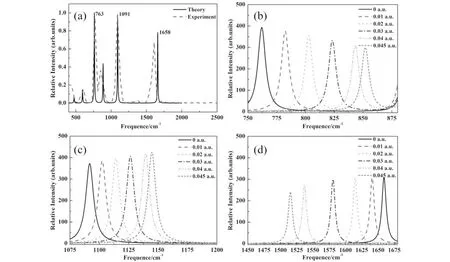
图2 1,1-二氯乙烯分子的红外光谱.(a) 无外电场作用下分子的红外光谱.实线表示理论计算得到的结果,虚线表示美国国家标准局(NIST)数据库提供的数据.(b)-(d) 不同外电场下763 cm-1、1091 cm-1和1658 cm-1的红外光谱Fig.2 The IRspectrum of 1,1-dichloroethylene (a) The IR spectrum in the absent of external electric field. solid line curve represents the theoretical results, and the dashed line represents the experimental results acquired from the database of National Institute of Standards and Technology. (b)-(d) the IR spectrum of 763 cm-1, 1091 cm-1 and 1658 cm-1 at different external electric field
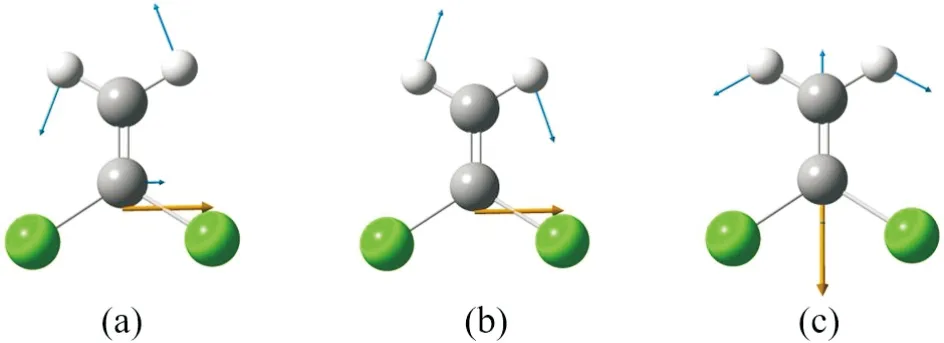
(a) 763 cm-1, (b) 1091 cm-1, (c) 1658 cm-1图3 1,1-二氯乙烯分子中三个主要的振动模式Fig.3 The three major vibrational modes in 1,1-dichloroethylene
2.4 不同外加电场对1,1-二氯乙烯分子激发态的影响
利用TD-DFT方法, 在B3LYP/6-311++G(d,p)高精度基组水平上进一步计算了1,1-二氯乙烯分子前8个激发态的激发能、激发波长以及紫外吸收光谱.表4和表5分别给出了不同外电场作用下前8个激发态的激发能和激发波长, 数据表明,随着外电场强度的增加,激发态能量逐渐减小,相应的激发波长逐渐从紫外变化到红外,这意味着在外加强电场作用下,分子更容易被激发而发生解离,这与2.2节得到的结论是一致的.

表4 不同外电场作用下1,1-二氯乙烯分子前8个激发态的激发能Tab.4 Excited energy of the first eight excited states of 1,1-dichloroethylene in different external electric field
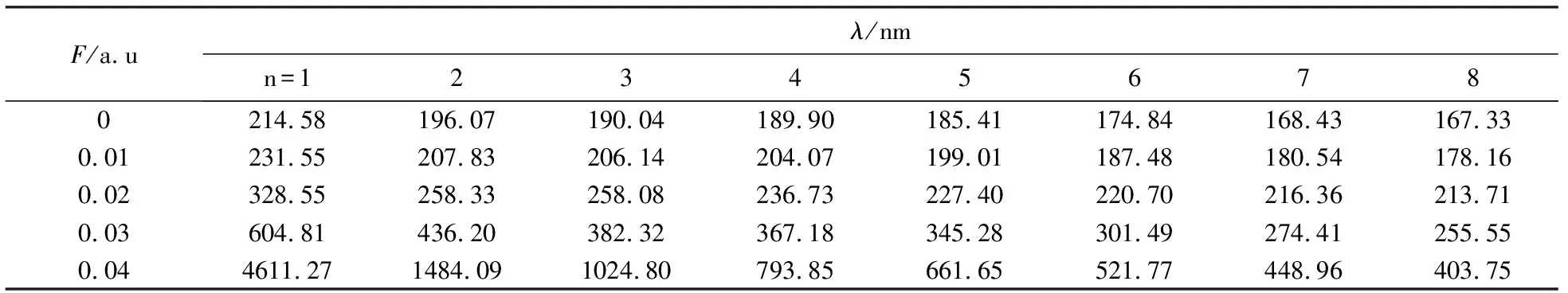
表5 不同外电场作用下1,1-二氯乙烯分子前8个激发态的波长Tab.5 Wavelengths of the first eight excited states of 1,1-dichloroethylene in different external electric field
利用相同的方法得到了1,1-二氯乙烯分子的紫外吸收光谱.如图4所示,在无外加电场的情况下,分子在紫外区域主要存在三个较强的吸收,分别对应的波长为196.25 nm,167.18 nm和141.95 nm,其中最强峰对应的波长为196.25 nm,这与TEEGAND等人在实验上观察到的结果是一致的[16],该吸收峰可以归属为C=C双键的π-π*跃迁,而167.18 nm和141.95 nm的紫外吸收则归属为分子里德堡的跃迁[16].有趣的是,随着C=C键正方向上电场强度增大,紫外吸收光谱呈现出明显的红移,这可能是由于外电场作用下电子云的离域扩展而导致共轭双键π-π*跃迁能量收缩造成的[17].
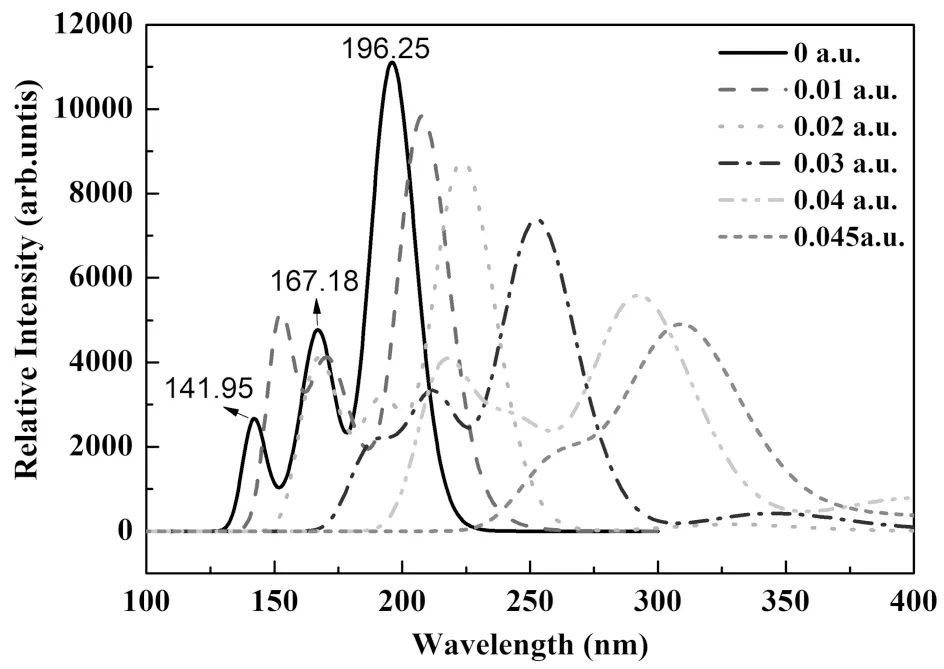
图4 不同外电场作用下1,1-二氯乙烯分子的紫外吸收光谱Fig.4 The UV absorption spectrum of 1,1-dichloroethylene in different external electric field
3 结语
本文利用Gaussian 03软件计算了1,1-二氯乙烯分子在外电场作用下的结构和光谱.首先获得了不同外电场对1,1-二氯乙烯分子键长、总能量、电偶极矩、电荷分布和轨道能级的影响.随着电场的增大,C=C键增长,C=Cl键缩短,分子的总能量升高,电偶极矩和分子的轨道能量则逐渐减小.其次,在分子优化构型的基础上,进一步获得了不同外电场作用下1,1-二氯乙烯分子的基态振动模式和红外光谱,结果表明,随着外电场的增加,C2H2基团摇摆振动频率呈现蓝移,而C=C伸缩振动模频率则呈现红移.最后,获得了不同外电场下分子激发态的能量、激发波长以及紫外吸收光谱,理论表明,随着外电场的增加,激发态能量逐渐减小,紫外吸收光谱呈现出了明显的红移现象.
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
原子与分子物理学报(2022年3期)2022-03-05
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
科技视界(2019年27期)2019-11-05
青岛大学学报(工程技术版)(2019年2期)2019-09-10
劳动保护(2018年8期)2018-09-12
恋爱婚姻家庭·养生版(2018年7期)2018-08-10
恋爱婚姻家庭(2018年21期)2018-07-23
中学化学(2015年8期)2015-12-29
