
WT1基因检测时机对Wilms肿瘤合并慢性肾脏疾病预后影响的回顾性队列研究
2019-12-19 06:32方晓燕毕允力汤小山刘佳璐张致庆翟亦晖李国民吴冰冰钱琰琰
中国循证儿科杂志 2019年4期
方晓燕 沈 剑 沈 茜 毕允力 汤小山 刘佳璐 张致庆 翟亦晖 陈 径 李国民 吴冰冰 钱琰琰 徐 虹 饶 佳
WT1(wilms tumor suppressor gene 1)是一种肿瘤抑制基因,在泌尿生殖系统的发育和Wilms肿瘤的发生发展中具有重要的意义[1,2]。WT1基因突变可以常染色体显性遗传,引发一系列肾小球疾病,包括Denys-Drash综合征(DDS)、Fraiser综合征(FS)和孤立型肾病综合征(INS)[1~4]。越来越多的研究显示基因分子诊断对WT1突变导致的Wilms肿瘤、肾病分型和预后有着重要的指导意义。
目前中国尚缺乏对WT1基因突变导致Wilms肿瘤和肾病的中国人群的基因型突变谱的大样本报道,在一些病例报道中亦缺乏长时间随访的资料,尤其是对WT1基因突变的肾病患者长期随访中发生肿瘤的风险及Wilms肿瘤术后发生慢性肾脏疾病(CKD)的预测和随访缺乏临床证据。本研究回顾性收集上海市肾脏发育与儿童肾脏病研究中心(中心)WT1基因突变患儿的临床症状和体征,分析在Wilms肿瘤合并CKD患儿中WT1基因检测对诊断和长期预后的影响。
1 方法
1.1 研究设计 以明确WT1基因突变为时间节点,以进展为终末期肾衰竭(ESRD)为终点事件,探讨基因检测时间节点前与后进展为ESRD对临床诊断分型和延缓进入ESRD病程的意义。
1.2 纳入标准 中心儿童肾脏病基因检测数据库中2001年1月1日至2018年12月31日明确WT1基因突变后临床随访>3个月且年龄<18岁的患儿,或Wilms肿瘤合并CKD 2~5期[7]、或SRNS、或有蛋白尿。
1.3 排除标准 已证实的继发性肾病综合征(NS),如各类病原体(HBV、HCV、HIV、梅毒)感染所引起的NS,如过敏性紫癜、系统性红斑狼疮、血管炎等系统性疾病引起的NS。
1.4 分组 明确WT1基因突变前和后进展为ESRD分别为早诊断组和晚诊断组。
1.5 随访治疗方案 ①确诊WT1基因突变之后,临床上减停就诊前已应用的激素或免疫抑制剂及其他有损害风险药物,予口服血管紧张素转化酶Ⅱ抑制剂为主的支持治疗;②Wilms肿瘤患儿根据肾功能调整化疗方案[4];③每年随访腹部超声筛查Wilms肿瘤的发生。
1.6 临床资料的采集 符合本文纳入和排除标准的病例进一步查询医院电子病案系统,采集发病年龄、首发症状、性别(包括染色体分析及表型性别)、出生史、母亲妊娠史、家族史、肾组织病理学改变、基因型的确定、激素治疗效应、是否进展为ESRD、从发病至进展为ESRD的病程或至随访终点(2018年12月12日)的病程、是否肾移植、肾外症状等临床资料。
1.7 基因检测
1.7.1 知情同意 行基因检测前获得患儿家长书面知情同意,对在患儿中发现的突变位点,得到监护人同意情况下在其家系中验证。
1.7.2 测序及分析 基因检测先后在北京智因东方转化医学研究中心/北京全谱医学检验实验室和复旦大学附属儿科医院分子诊断中心进行。用EDTA抗凝管采集外周血2 mL,采用QIAGEN公司(德国)的血液基因组DNA提取试剂盒抽提外周血基因组DNA,分别采用Sanger测序、Snapshop技术及WES技术行基因分型。直接测序应用基因组DNA分别扩增候选基因外显子及其侧翼序列,扩增引物参照已报道的文献[3],使用ABI3730DNA分析仪(美国ABI公司)行双向直接测序分析。WES采用外显子捕获的方法对4 000种人类单基因病的相关致病基因进行高通量测序,利用生物信息学对测序结果进行分析,用Sanger法对高通量测序结果进行家系验证。依据WT1测序结果分析,由第9内含子区域上的剪切供体位点突变诱发,从而导致第9外显子编码的赖氨酸-苏氨酸-丝氨酸(简称KTS)组成的三肽片段插入或缺失,形成+KTS/KTS两种蛋白亚型,据此定义KTS突变。
1.8 统计学分析 分类资料应用Fisher确切概率法(病例数不满足卡方检验要求),定量资料应用Mann-whitney检验;应用Prism 8软件作图。
2 结果
2.1 一般情况 符合本文纳入和排除标准,确诊为WT1基因突变致蛋白尿、或NS、或Wilms肿瘤合并CKD患儿22例患儿进入本文分析。表1显示,早诊断组和晚诊断组各11例,两组在生物学性别、起病月龄、临床分型(DDS/FS/NS)、WT1基因突变型(错义突变/KTS突变/剪切位点突变)、Wilms肿瘤发生和临床应用化疗的病例数差异均无统计学意义。早诊断组和晚诊断组在WT1基因确诊前分别有2例(18.2%)和8例(72.7%)使用激素或免疫抑制剂治疗,差异有统计学意义。
2.2WT1基因突变分析 表2显示,在22例患儿中发现了WT1基因12种不同的单一杂合突变,所有的突变均位于第8~9外显子/内含子。3例患儿中分别检测到位于第9内含子剪切位点的KTS突变[3](NM_024426.4: c.1432+4C>T; c.1432+5G>A),其中早诊断组1例,晚诊断组2例。
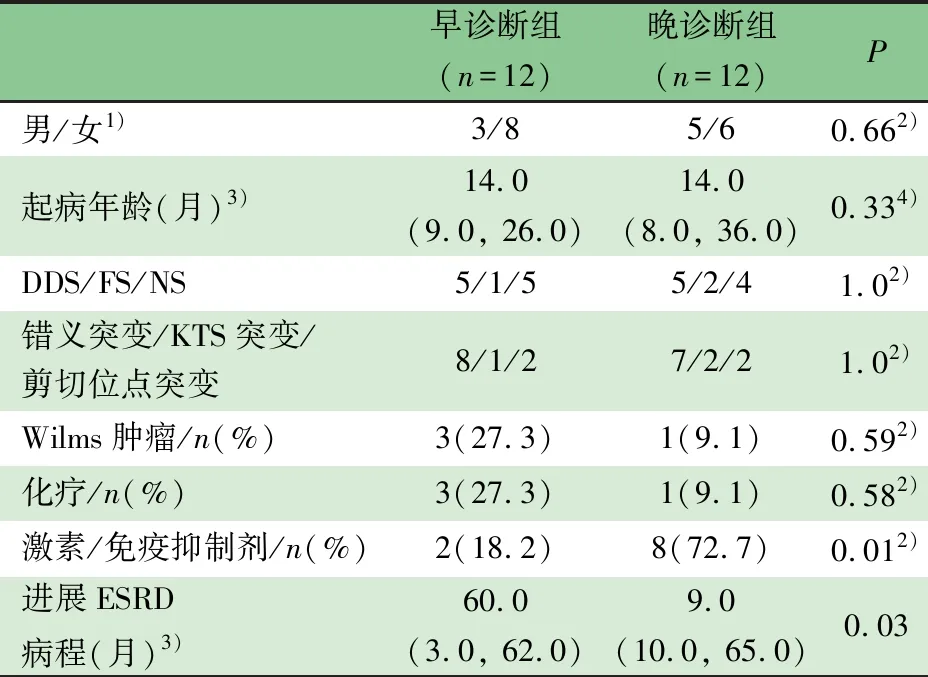
表1 WT1基因突变相关慢性肾脏疾病患者中早诊断组与晚诊断组的临床特征
注 DDS:Denys-Drash综合征,ESRD:终末期肾衰竭,FS:Fraiser综合征,NS:孤立型肾病综合征;1)生物学性别;2)Fisher exact test检验; 3)中位数(P25,P75);4)Mann-whitney检验
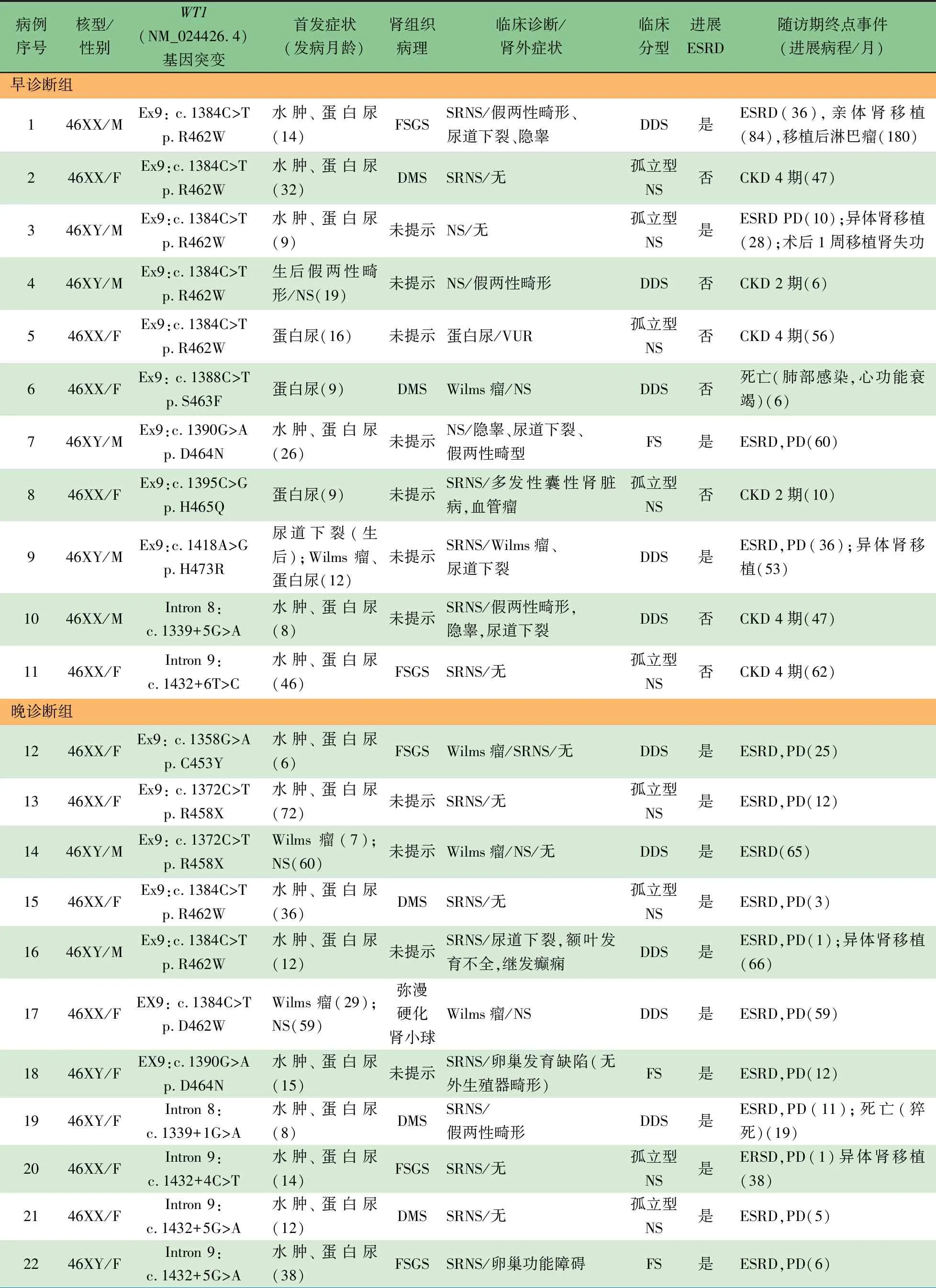
表2 22例WT1基因突变合并慢性肾脏疾病患儿的基因型和表型
注 CKD:慢性肾脏疾病;DDS:Denys-Drash综合征;DMS:弥漫性系膜硬化性肾小球肾炎;ESRD:终末期肾衰竭;FS:Fraiser综合征;FSGS:局灶节段性硬化肾小球肾炎;M:男;F:女;NS:肾病综合征;PD:腹膜透析;SRNS:激素耐药性肾病综合征;VUR:膀胱输尿管返流12例患儿中分别诊断了已报道的位于第9外显子上的错义突变(NP_077744.3: p.R458X,p.R462W和p.D464N)[3];同时发现了8个新发突变(NM_024426.4: c.1339+1G>A, c.1339+5G>A, c.1339+1G>A, c.1432+6T>C, p.C453Y,p.S463F, p.H465Q, p.H473R)。位于第8内含子的两个剪切位点突变(c.1339+1G>A和c.1339+5G>A)及位于第9内含子的剪切位点突变(c.1432+6T>C),经计算机软件分析工具Human Splicing finder预测为显著可能的剪切位点。
2.3 临床表型特征分析 10例患儿表型为DDS,3例为FS,9例为INS,至随访终点尚未发现Wilms瘤的复发;3种临床分型在早诊断组和晚诊断组中比例差异无统计学意义。根据核型分析判断9例为男性,其中3例核型判定为46XY,而表型呈现女性外生殖器的假两性畸形;2例核型判断为46XX,而表型呈现男性外生殖器的假两性畸形。假两性畸形在早诊断组中4例,晚诊断组中1例。9例进行了肾穿刺病理检查,其中5例为弥漫性系膜硬化性肾小球肾炎(DMS),5例为局灶性节段性肾小球硬化(FSGS),1例为弥漫硬化性肾炎(表2)。
2.4 肾功能进展恶化及透析移植随访情况分析 图1显示,22例患儿在随访终点进入ESRD有15例,其进展为ESRD的病程1~65个月。早诊断组至随访终点4例进入ESRD,4例进入CKD 4期,1例进入CKD 3期,2例进入CKD 2期;2例成功完成了肾移植。晚诊断组中11例进展至ESRD,均进行腹膜透析治疗,其中2例成功完成了肾移植。应用生存曲线分析比较发现(图2),早诊断组进展至ESRD病程的中位数为60.0个月,晚诊断组进展至ESRD病程的中位数为9.0个月,差异有统计学意义(P=0.011)。
在晚诊断组中,2例首发单纯Wilms瘤患儿在单侧肾切除术及化疗后,随访发现蛋白尿,肾功能已进展为ESRD;1例生后即存在尿道下裂发育缺陷,1岁龄发现NS,在1个月内进展为ESRD,后完善基因检测明确WT1致病突变,确诊DDS。在早诊断组中,3例NS后蛋白尿起病,病初就诊后发现Wilms瘤,行单侧肾脏切除术及化疗,随访中均进入ESRD。早诊断组1例患儿在1岁龄以蛋白尿起病无合并其他肾外症状,肾穿刺病理发现肿瘤细胞浸润,诊断Wilms瘤,后随访中因肺部感染,心功能衰竭死亡。4例进行了肾移植,1例在移植前预防性行双肾切除术,4例移植手术同期进行了残余肾的切除,并完善病理未发现肿瘤复发。4例均未发生排斥反应或移植肾失功,其中1例亲体肾移植在随访8年发现了肝脏的淋巴瘤病灶,化疗缓解中,其余3例移植后至本次研究随访终点未发生肿瘤复发或新生肿瘤。
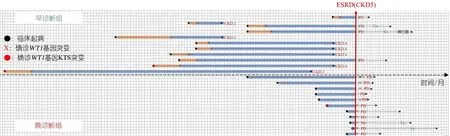
图1WT1基因突变相关慢性肾脏疾病患者中早诊断组与晚诊断组的临床进程
注 ESRD:终末期肾衰竭;PD:腹膜透析;Tx:肾移植。早诊断组(虚线以上部分)至随访终点,进展为CKD 2期、3期和4期分别为2、1和4例,4例进展为ESRD(CKD 5期),后行腹膜透析(PD)治疗,有2例成功完成了肾移植(Tx)。晚诊断组(虚线以下部分)均行PD治疗,2例成功完成了Tx
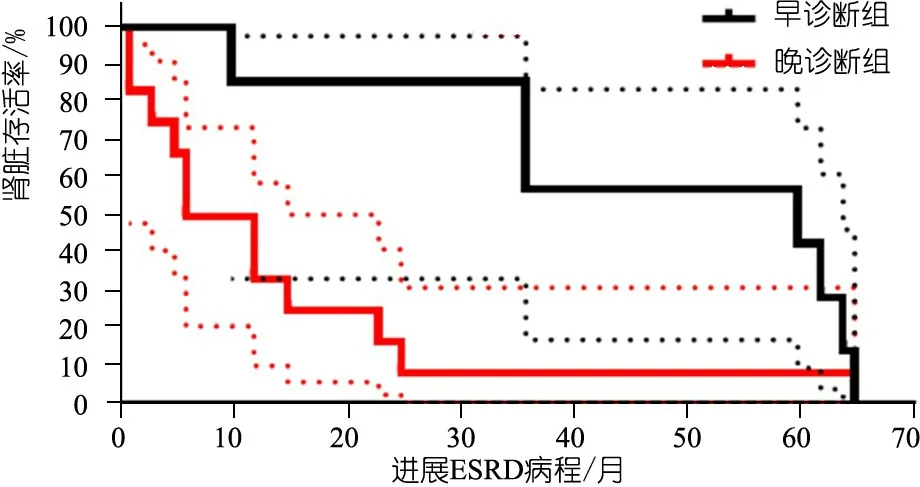
图2WT1基因突变早诊断组和晚诊断组进展至ESRD病程间隔的Kaplan-Meier生存曲线分析
注 早诊断组与晚诊断组患儿比较进展为ESRD的病程间隔(月)差异有统计学意义(P=0.011);晚诊断组进入ESRD病程更短,Hazard Ratio(Logrank) 0.42(95%CI, 0.17~1.0)
3 讨论
WT1基因首先在肾母细胞瘤(又称Wilms瘤)中作为抑癌基因被克隆鉴定,作为首个被发现证实的儿童NS的致病基因,开启了人们对肾病的遗传分子学研究。WT1基因位于染色体11p13,编码蛋白包括锌指蛋白DNA结合区参与转录后调控及RNA识别。单纯Wilms肿瘤患者致病突变集中在WT1基因的N端第1-5外显子区域,NS合并性腺发育障碍或Wilms瘤患者的致病突变报道集中在C端第6-9外显子区域[8]。WT1基因作为最常见的先天性NS的致病基因之一,约7%儿童NS筛检出WT1致病突变,在不同地区报道的明确致病基因的儿童NS中比例17%~50%[3,4,6]。在儿童NS中行WT1基因的检测有助于明确临床分型及判断预后。本研究依据WT1基因突变诊断明确时间节点与进展为ESRD终点事件的先后,从临床发病年龄、肾脏病理、进展至ESRD病程间隔时间、核型及基因型分析、肾外症状及长期随访情况等,全面比较了早诊断组与晚诊断组的临床特征及预后,提示在ESRD之前明确WT1基因突变型对长期延缓肾功能恶化的临床意义。
本研究中早诊断组患儿,在起病初期发现蛋白尿合并Wilms肿瘤或假两性畸形等其他肾外症状,临床上怀疑WT1基因相关综合征,及早完善了基因检测。晚诊断组患儿,病初仅发现Wilms瘤,或仅蛋白尿起病,未发现其他肾外症状,在进展至ESRD后完善了基因检测,晚诊断组比早诊断组患儿有更快进展为ESRD趋势。尽管临床分型包括DDS、FS及INS在早诊断组和晚诊断组分布比例差异无统计学意义,但本研究中尚缺乏对早诊断和晚诊断的影响因素进行进一步分析。许多患儿由于缺乏肾病起病之前的尿检筛查,不能排除生后即有蛋白尿而未能及早诊断,以至肾功能迅速恶化。在晚诊断组中蛋白尿或NS起病患儿,往往在未明确基因诊断之前先后应用了较长疗程的激素及免疫抑制剂,药物对肾小管间质的损害有可能参与了肾功能的加速恶化。来自欧洲的肾脏病登记数据库显示,诊断WT1第8或第9外显子及内含子区域的致病突变的42例患者,40.5%合并Wilms瘤,83.3%进展至ESRD[9]。因此,临床发现Wilms瘤患者须及早明确WT1基因的分子诊断,对判断预后及合理制定治疗方案,保护残存肾功能具有重要意义。
在晚诊断组中,2例首发症状为Wilms肿瘤而无蛋白尿起病,经单纯肾切除术及化疗后,分别在发现肿瘤后随访2.5年及随访4.1年发现蛋白尿,进展为ESRD。提示临床医生在Wilms瘤患者随访中重视尿液检查;同时早期明确基因诊断,如明确WT1基因第8或9外显子及内含子突变,则须考虑诊断WT1相关综合征DDS或FS,术后及化疗后必须重视肾功能的保护和随访。1例生后发现尿道下裂,首次就诊发现NS,肾功能急进恶化,病程1个月进入ESRD,转入中心行腹膜透析治疗后明确了WT1基因诊断。提示尿道下裂出生缺陷在诊断多系统疾病的鉴别中需要引起重视。
WT1基因突变为杂合胚系突变,尽管大部分DDS或FS男性患儿存在严重的生殖器官发育异常伴条索状性腺和不育,但多数DDS或FS女性患儿具有正常的生殖器官,对患儿长期性腺功能的评估随访及心理评估尚需进一步加强[10]。本研究中5例假两性畸形患儿均合并尿道下裂,其中3例合并隐睾,2例合并卵巢发育缺陷。因此,对外生殖器发育缺陷的患儿应及早进行基因检测,同时重视尿液检查的随访。对携带WT1基因突变患者的核型分析及性腺功能科学评估对患儿的长期生活质量的提高意义重大。
综上所述,针对儿童Wilms肿瘤、NS或蛋白尿、先天性外生殖器发育缺陷患儿中,在肾功能进展恶化之前及早明确WT1基因突变分析,不仅有助于临床诊断分型,还能显著延缓进入ESRD病程。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
昆明医科大学学报(2021年12期)2021-12-30
老友(2021年3期)2021-03-28
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
家庭百事通·健康一点通(2020年11期)2020-11-30
肿瘤预防与治疗(2019年6期)2019-07-30
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
家庭医药·快乐养生(2016年4期)2016-05-11
