
全羧化酶合成酶基因新发突变致以皮肤为首发症状的多种羧化酶缺乏症一例
2019-12-19 05:53李云玲郑惠文李寅王丽华李薇郭小璇黄春兰周沙黄新文吕中法
中华皮肤科杂志 2019年11期
李云玲 郑惠文 李寅 王丽华 李薇 郭小璇 黄春兰 周沙 黄新文 吕中法
1浙江大学医学院附属儿童医院皮肤科,杭州 310052;2杭州大关上塘社区卫生服务中心皮肤科 310026;3 浙江大学医学院附属儿童医院遗传与代谢科,杭州310052;4浙江大学医学院附属第二医院皮肤科,杭州 310009
多种羧化酶缺乏症(multiple carboxylase deficiency,MCD)是一种常染色体隐性遗传、代谢性疾病,可表现为皮肤、神经、呼吸、消化等多系统的非特异临床症状,诊治不及时可导致死亡和中枢神经系统不可逆损伤[1-4]。本文报道1例以皮肤为首发症状的多种羧化酶缺乏症及新发突变。
一、病例资料
患儿男,3个月12天,因皮疹2个月,气促伴哭吵3 d入院。患儿自出生后1个月起阴囊、会阴、臀部和肛周皮肤出现边界清楚的红斑伴鳞屑(图1),逐渐口周、腋下、肘部屈侧、腘窝及颈部也出现上述皮疹,蔓延分布全身。皮损处无明显瘙痒,非皮损区皮肤无明显干燥、脱屑。在多家医院诊断为湿疹,给予外用糖皮质激素、联苯苄唑等软膏治疗无明显缓解。后又诊断为肠病性肢端皮炎,给予口服葡萄糖酸锌治疗10 d 无好转。入院前3 天出现气促,清醒及睡眠时均明显,伴哭吵烦躁、呕吐,吐出物为奶汁样胃内容物,非喷射状。病程中无抽搐,近日无发热及咳嗽。患儿为第3 胎第3 产,足月剖宫产,母乳喂养。出生体重3.2 kg,Apgar 评分正常。父母非近亲结婚,否认家族遗传病史,家族无类似疾病患者。
体检:呼吸 58 次,血压 83/37 mmHg(1 mmHg = 0.133 kPa),体温36.6 ℃,脉搏152 次。神清,反应差,前囟平软,体重6.6 kg,头颅五官无畸形,中度三凹征,无紫绀,无喘鸣,双肺无啰音,心音有力,心律齐,无杂音。腹软,肝脾肋下未触及,肠鸣音正常。四肢肌张力及肌力正常。皮肤科检查:全身皮肤潮红,伴糠秕状或小片状干燥鳞屑,部分浅糜烂伴浆液性结痂,无丘疹、水疱及脓疱。头发、眉毛、睫毛无脱落。
实验室检查:血气分析pH 7.08(参考值7.35 ~ 7.40,下同),乳酸12.2 mmol/L(1.5 ~ 1.6 mmol/L),血氨103 μmol/L(9 ~ 30 μmol/L)。血常规白细胞14.61 × 109/L,淋巴细胞0.55,中性粒细胞0.30,红细胞平均血红蛋白浓度135 g/L,血小板 125 × 109/L,C 反应蛋白 7 mg/L。血糖 6.1 mmol/L(3.6 ~ 6.1 mmol/L),血钾3.0 mmol/L(3.5 ~ 5.5 mmol/L),血锌水平正常。尿酮阳性(+++)。肝肾功能、甲状腺功能、肾上腺皮质激素、免疫球蛋白补体、细胞免疫功能、细胞因子检查无异常。腹部B 超未见异常。胸部X 线摄片:双肺纹理增多,心影未见明显增大,形态位置无特殊,两膈光整,肋膈角锐利。头颅磁共振检查正常。心电图、心脏彩超正常。双肾输尿管、肝、脾、胆、胰腺B超未见异常。
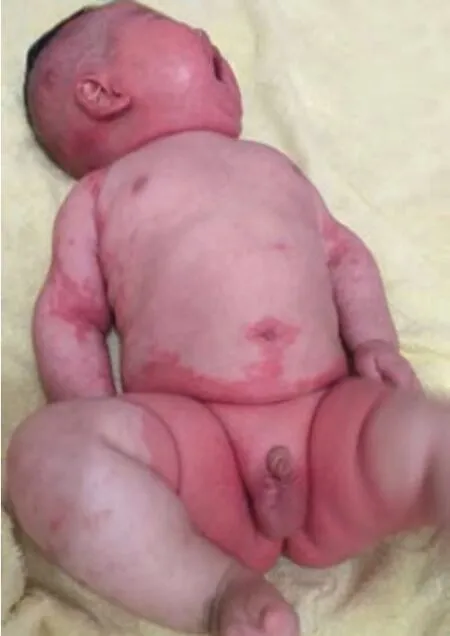
图1 患儿肘窝、阴囊、会阴见红斑、糠秕样鳞屑
二、诊断经过
患儿以湿疹伴感染、代谢性酸中毒收入院。入院后给予美罗培南抗感染,2.5%碳酸氢钠纠正酸中毒,同步间歇指令通气辅助通气,3 d 后代谢性酸中毒纠正并脱离呼吸机,但血乳酸(7.4 ~ 10.8 mmol/L)、血氨(50 ~ 88 μmol/L)仍高。血串联质谱检测结果显示,丙酰基肉碱4.33 μmol/L(0.43 ~3.8 μmol/L),甲基丙二酰基肉碱+ 3 羟基异戊酰基肉碱20.12 μmol/L(0.07 ~ 0.4 μmol/L),甲基丙二酰基肉碱 +3 羟基异戊酰基肉碱/丙酰基肉碱 4.65 μmol/L(0.05 ~0.39 μmol/L),甲基丙二酰基肉碱+3羟基异戊酰基肉碱/辛酰基肉碱 503 μmol/L(0.65 μmol/L ~ 10 μmol/L)。尿气相色谱质谱检测显示,除乳酸、酮体明显增高外,多种有机酸(3 甲基巴豆酰甘氨酸、3 羟基异戊酸、3 羟基丙酸、柠檬酸)也明显升高。患儿存在难以纠正的高乳酸血症、高血氨及有机酸尿症,考虑多种羧化酶缺乏症。给予口服5 mg生物素每日3 次,治疗3 d,血乳酸(1.2 mmol/L)、血氨(18 μmol/L)、尿酮恢复正常,全身红斑、脱屑好转。
采用二代测序及Sanger 测序验证显示,患儿存在全羧化酶合成酶基因(holocarboxylase synthetase,HLCS)第9 外显子1522处胞嘧啶>胸腺嘧啶(c.1522C>T)和第11外显子1796_1814 缺失(c.1796_1814del)突变,患儿母亲及父亲分别发现HLCS基因c.1796_1814del及c.1522C>T突变(图2)。
诊断:MCD。
三、治疗
根据患儿血、尿有机酸指标调整生物素剂量,目前给予生物素5 mg每日2次长期维持治疗。随访1年,患儿病情稳定,血、尿有机酸和血氨正常。患儿体格发育、神经和精神发育正常,运动和语言能力发育正常,听力正常。头发正常,无脱发。脑电图正常。
四、讨论
MCD 如能早期确诊,给予生物素治疗,预后良好[2]。HLCS 基因位于21q22.13,由12 个外显子组成,共编码726 个氨基酸,HLCS 基因突变导致全羧化酶合成酶活性下降,影响生物素与生物素依赖的多种羧化酶结合,导致生物素相对不足,从而影响多种羧化酶的活性[1-2,5]。由于缺乏多种羧化酶,机体线粒体依赖生物素的4种羧化酶(丙酮酸羧化酶、丙酰辅酶A 羧化酶、3 甲基巴豆酰辅酶A 羧化酶及乙酰辅酶A 羧化酶)活性下降,机体脂肪酸合成,糖异生和氨基酸酸代谢障碍,有机酸等异常代谢产物在血、尿中蓄积,导致发育迟缓、共济失调、癫痫等神经系统症状及皮炎、代谢性酸中毒等[1,3,6]。
本例患儿以皮肤红斑、鳞屑为首发症状,皮损无明显瘙痒,外用糖皮质激素治疗无改善,病程中出现气促、代谢性酸中毒、高乳酸血症、高氨血症、高有机酸尿。基因检查显示,患儿HLCS 外显子区域存在c.1522C>T 和c.1796_1814del 两处突变,从基因水平证实为MCD,突变基因分别来自父母。c.1522C>T位点为已知致病突变位点[7],为错义突变,导致第508 位上的精氨酸被色氨酸取代;c.1796_1814del 突变为移码突变,导致第599 位的甘氨酸被缬氨酸取代,并且从该位点往后第24 个密码子变为终止密码子,导致翻译停止,蛋白截短。我们在已发表的ExAC(Exome Aggregation Consortium)、GnomAD (Genome Aggregation Database)、ClinVar 和 HGMD (Human Gene mutation Database)数据库中未发现HLCS 基因有c.1796_1814del 突变的报道,该位点为新发突变致MCD,突变导致的蛋白截短对全羧化酶合成酶影响大,依据美国医学遗传学与基因组学学会指南,该新发突变评级为致病突变。
本文报道1 例典型早发型多种羧化酶缺乏症,通过血串联质谱和尿气相色谱质谱检测有机酸及基因测序得以确诊,确诊后给予生物素治疗疗效佳,病情稳定,血、尿有机酸和血氨维持正常。
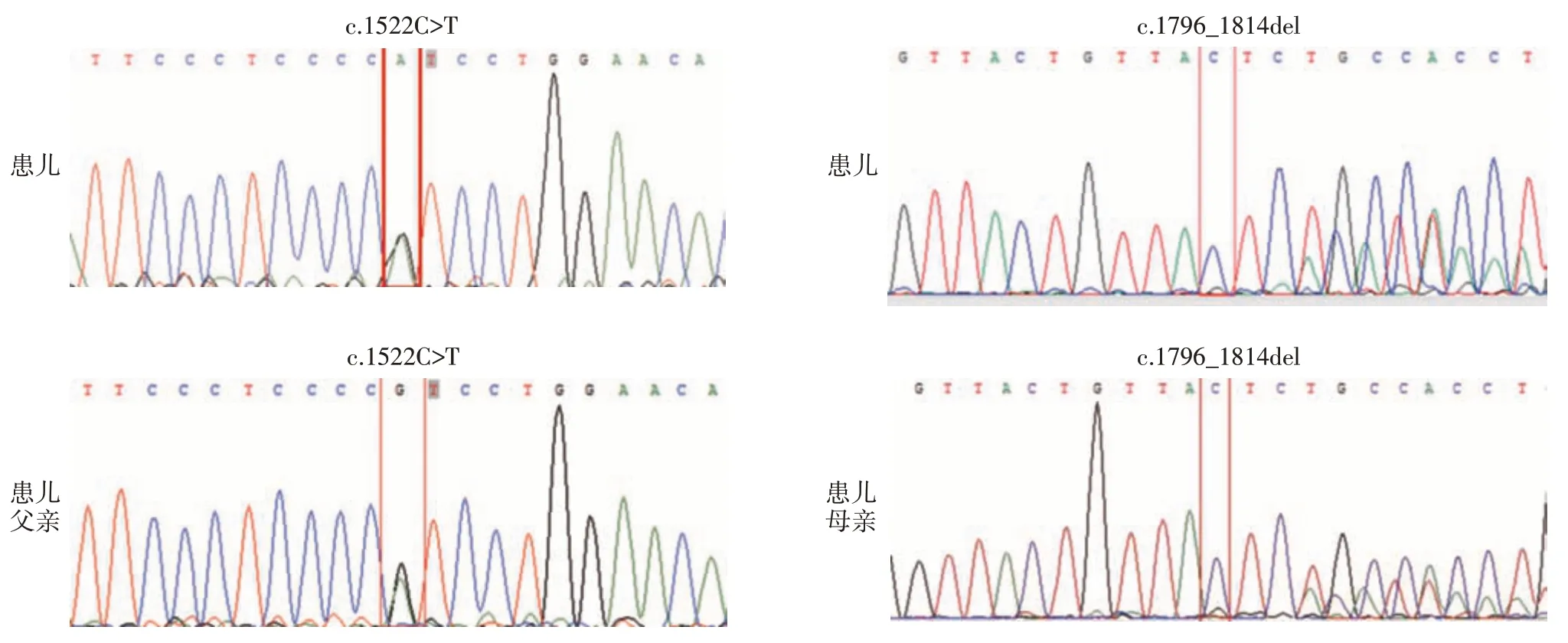
图2 患儿及其父母全羧化酶合成酶基因(HLCS)测序图 患儿HLCS 基因第9 外显子c.1522C>T(胞嘧啶>胸腺嘧啶)和第11 外显子c.1796_1814del(缺失突变)存在复合突变;患儿父亲存在c.1522C>T突变,母亲存在c.1796_1814del突变
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
现代检验医学杂志(2021年3期)2021-12-03
现代检验医学杂志(2021年2期)2021-04-17
农业工程学报(2021年3期)2021-04-15
兽医导刊(2020年23期)2020-12-30
茶叶(2020年1期)2020-05-30
山东农业科学(2019年9期)2019-12-09
无机化学学报(2019年2期)2019-02-27
河北渔业(2017年6期)2017-06-30
中国实用医药(2016年19期)2016-08-05
天津农业科学(2015年12期)2015-12-03
