
类Friedel-Crafts酰基化合成苯基二氯化膦中催化剂AlCl3的催化与失活机理探究
2019-11-29 07:49段好东2刘大勇3曹迁永4王忠卫
山东科技大学学报(自然科学版) 2019年6期
郭 阁,段好东2,刘大勇3,曹迁永4,王忠卫
(1. 山东科技大学 材料科学与工程学院,山东 青岛 266590;2. 山东科技大学 化学与环境工程学院,山东 青岛 266590;3. 黄淮学院 化学与制药工程学院,河南 驻马店 463000;4. 南昌大学 化学学院,江西 南昌 330031)
苯基二氯化膦(dichlorophenylphosphine,BPD)是一种重要的有机磷化合物,工业上被广泛用于合成阻燃剂、光引发剂、医药等含磷化学品。大量文献研究报导了BPD的合成方法,如离子液体合成法[1]、分子筛负载合成法[2]、三苯基膦合成法[3]等,上述方法均难以实现工业化[4]。BPD的工业合成方法主要基于两类反应:一类是Michaelis提出的苯和PCl3在无水AlCl3作用的类Friedel-Crafts酰基化反应法[5];另一类是氯苯、PCl3和单质磷在催化剂无水AlCl3作用下的高温高压反应法[6]。目前,类Friedel-Crafts酰基化反应法是工业上采用最广泛的BPD合成方法。
类Friedel-Crafts酰基化合成BPD的反应与经典Friedel-Crafts酰基化反应有许多类似之处,主要表现在:①反应为芳香烃(苯)与亲电体(配合物PCl3-AlCl3)的亲电取代反应,副产物为HCl;②反应需要化学计量数的Lewis酸(AlCl3)为催化剂;③反应生成物与催化剂形成络合物,需要先解离络合物,再分离生成物;④AlCl3与生成物的络合导致AlCl3失活,不能进一步催化反应,从而使反应不具有循环催化的特点。
尽管有不少关于类Friedel-Crafts酰基化反应合成BPD的研究,但大多数开展的是BPD与AlCl3络合物的解离方式与BPD的分离方法研究,针对苯与PCl3在AlCl3催化下反应机理的研究则比较少。对反应机理的认识如图1(a)所示,假定PCl3在AlCl3的作用下形成[Cl2P]+[AlCl4]-离子配合物,[Cl2P]+[AlCl4]-或[Cl2P]+作为亲电试剂,进攻芳环生成BPD。但此机理存在较大的争议,至今并无实验依据来证实其中阳离子或配合物存在的文献报道。由于对所提出的反应机理缺乏实验证据,后来一些综述在描述此类反应时,也没有明确说明是[Cl2P]+[AlCl4]-或[Cl2P]+作为亲电试剂进攻苯环,进而发生亲电取代反应,而是更加谨慎地用“亲电进攻体结构不明确”来说明反应机理。
本课题组多年来一直致力于BPD的合成[7-8]及其衍生物的应用研究[9-11],发现:在AlCl3作用下,苯与PCl3反应除生成BPD外还会生成一定量的二苯基氯化膦(chlorodiphenylphosphine, DPC),即BPD与苯在AlCl3作用下可能进一步发生了如图1(b)所示的反应得到DPC与AlCl3的络合物,而DPC与苯则无法进一步反应生成三苯基膦(triphenylphosphine,TPP)。这个现象引起了课题组的兴趣。在PCl3、苯、AlCl3反应生成BPD和DPC的反应中,AlCl3与PCl3、BPD和DPC形成配合物的配位方式和配位能力很大程度上决定了反应能否进行。为了解释产生这个现象的原因,同时揭示苯与PCl3在AlCl3作用下的类Friedel-Crafts酰基化反应机理,采用量子化学的研究方法,通过配合物分子表面的静电势分析、两单体之间的相互作用能、分子中原子的量子理论(quantum theory of atoms in molecules,QTAIM)、定域化轨道指示函数(localized orbital locator,LOL)和独立梯度模型(independent gradient model,IGM)等分析方法,研究PCl3、BPD、DPC分别与AlCl3的配位方式和配位能力,期望为芳烃与PCl3的反应研究提供参考。
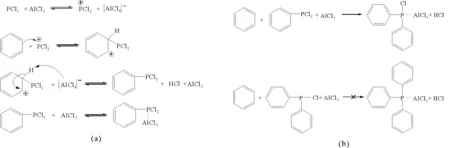
图1 AlCl3催化类Friedel-Crafts酰基化反应制备BPD反应机理图Fig. 1 Reaction mechanism of similar Friedel-Crafts acylation diagram of the BPD formation catalyzed by AlCl3
1 理论计算方法
利用密度泛函理论,在B3LYP/6-31G(d,p)[12]计算水平上对单体分子(AlCl3、PCl3、BPD和DPC)进行结构优化,并分析了单体分子表面静电势(electrostatic potential analysis,ESP)分布,预测两单体的配位点,进而初步构造配合物分子模型。在B3LYP-D3/6-31+G(d,p)考虑DFT-D3色散作用的计算水平上,对所有的初始配合物分子进行构型优化及频率计算,得到无虚频且较稳定的构型。为了研究两单体分子之间的相互作用能,在M062X-D3/6-311+G(d,p)计算水平上进行配合物分子的单点能计算,在考虑基组重叠误差(basis set superposition error,BSSE)校正的基础上,两单体分子之间的相互作用能为:Einter=EA-B-EA-EB+EBSSE。通过Multiwfn程序[13]进行配合物分子表面静电势分析,两单体分子之间成键原子之间的(Cl—Al键或P—Al键)的键级、QTAIM、IGM和LOL分析,其中将产生的表面静电势和IGM的分析文件载入VMD程序绘制完成ESP分布图和IGM等值面为0.01 a.u.的分布图。以上计算均由Gaussian 09 程序[14]完成。
2 结果与讨论
2.1 分子表面静电势分析
2.1.1 两单体分子之间配位点的预测
通过Multiwfn程序计算分析单体分子(PCl3、BPD、DPC和AlCl3)的范德华表面静电势分布[15],图2是单体PCl3、BPD、DPC和AlCl3在电子密度为0.001 a.u.的等值面上的分子静电势图。分子表面的静电势分析能够预测两种单体之间的配位点:在PCl3、BPD和DPC中,P原子的正上方存在局部静电势极小值Vs,min,P,对应P原子上的存在的孤对电子;Cl原子周围为负静电势区,其中存在局部静电势极小值Vs,min,Cl;AlCl3中Al原子正上方具有静电势极大值258.78 kJ·mol-1;由于静电吸引作用,Al原子进攻三种单体的静电势较小的区域即可形成配合物。
表1所示为各种单体分子的表面静电势局部极小值数值。通过三种单体的静电势数值可以看出,随着给电子取代基苯环的增加,P原子和Cl原子周围的电子密度不断增加,静电势局部极小值数值更负。若三种单体与AlCl3形成配合物,那么配合物的相互作用强度会按照PCl3、BPD、DPC依次增强。对于同一种单体来说,P原子和Cl原子周围都具有静电势局部极小值,局部电子密度较大,都有可能成为AlCl3的配位点,而且Cl原子的Vs,min,cl要比P原子的Vs,min,P的数值要负,Cl原子更易与AlCl3形成配合物。由ESP分析得出AlCl3中Al原子(静电势极大值区域)进攻三种单体的静电势较小的区域(Cl原子的Vs,min,cl区域,P原子的Vs,min,P区域),可形成两种类型的配合物,将AlCl3中的Al原子与PCl3、BPD和DPC中的Cl原子配位形成的配合物定义为A型配合物;将AlCl3中的Al原子与PCl3、BPD和DPC中的P原子配位形成的配合物定义为B型配合物。根据以上静电势分析结果来搭建配合物初始结构,进而继续进行配合物的计算研究。
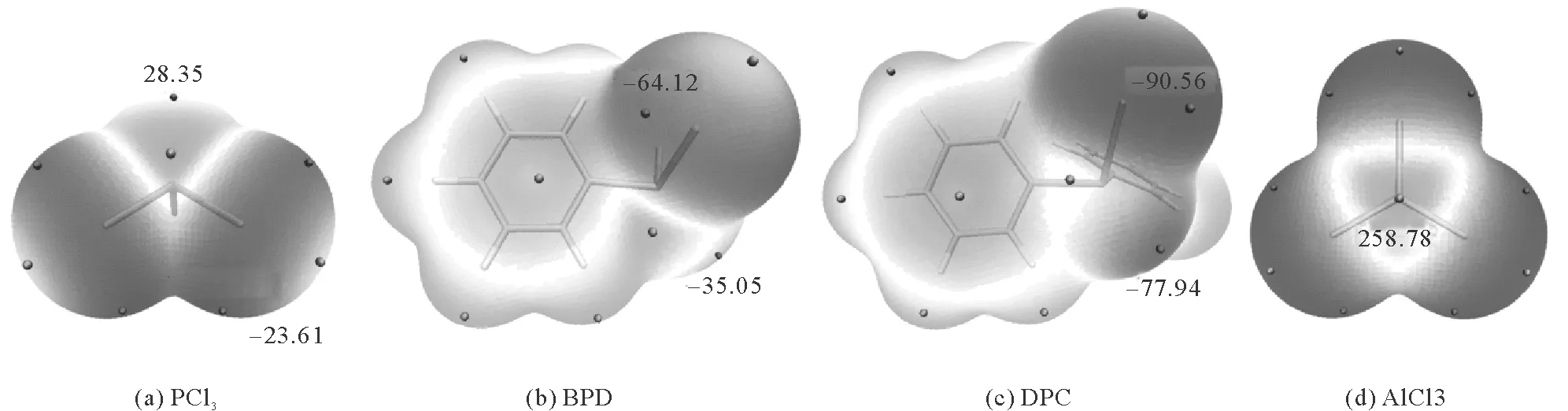
图2 在B3LYP/6-31G(d,p)计算水平上四种单体的表面静电势分布图Fig. 2 The electrostatic surface potential map of four monomers at the B3LYP/6-31G(d,p) level

表1 分子PCl3,BPD和DPC中Cl原子和P原子周围静电势的局部极小值(Vs,min)Tab. 1 Local minimum values of electrostatic potential around Cl and P atoms in molecules PCl3, BPD and DPC
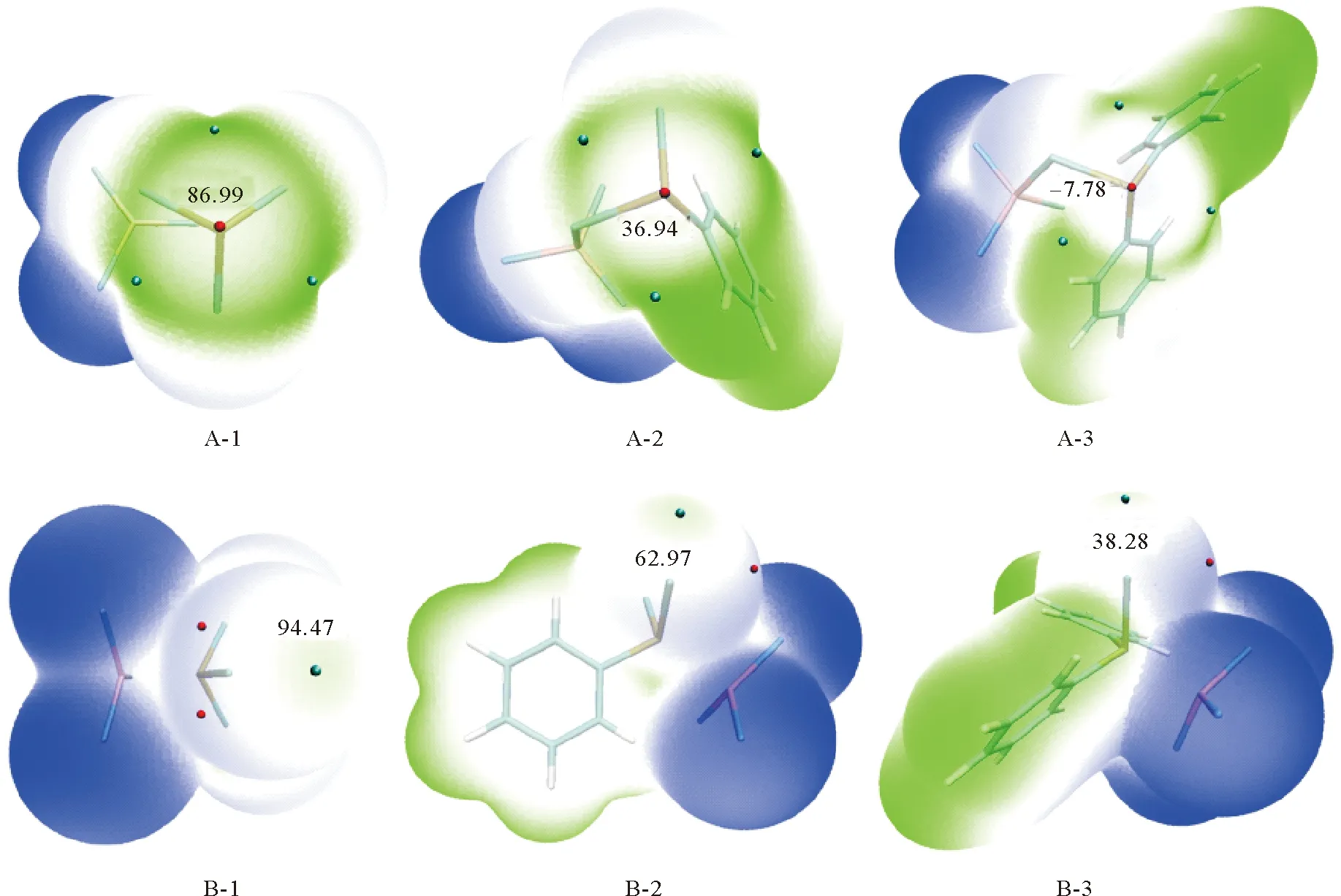
2.1.2 配合物的反应活性位点分析
在B3LYP-D3/6-31+G(d,p)计算水平上,对两种类型的配合物分子初始结构进行构型优化,得到较稳定的结构。在类Friedel-Crafts酰基化制备BPD的反应过程中,推测机理:主要是催化剂AlCl3与单体(PCl3和BPD)形成配位物,AlCl3夺取单体的电子形成P正离子,P正离子能够进攻苯环,从而发生亲电取代反应。为了研究两种类型的配合物在反应中的反应活性,通过配合物的静电势,可以分析预测反应活性位点,从而验证推测的反应机理。图3为两种配位类型的配合物分子的静电势分布图,其中给出了Vs,min,P和Vs,max,Cl的静电势数值。当AlCl3中的Al原子与PCl3、BPD、DPC中的Cl原子进行配位时,夺取单体的电子,使P原子上的电子密度减少,静电势数值增大。其中A-1 (A型PCl3-AlCl3)和A-2(A型BPD-AlCl3),P原子正上方的静电势极小值为正值,对苯环有进攻能力,可以进行亲电取代反应;而A-3(A型DPC-AlCl3)中P原子正上方的静电势极小值为负值,就无法再去进攻苯环生成TPP。当AlCl3中的Al原子与PCl3、BPD、DPC中的P原子进行配位时,虽然配合物中Cl原子上方出现静电势为正的极大值,但Cl原子进攻苯环的能力较弱;P原子被催化剂AlCl3所束缚,就很难进攻苯环发生反应。由以上配合物分子的ESP分析得出A型配合物的形成更利于反应的顺利进行。
2.2 配合物的相互作用能对比分析
对PCl3-AlCl3(1)、BPD-AlCl3(2)和DPC-AlCl3(3)配合物的配位成键特点和两单体分子之间的相互作用能进行研究。表2给出了配合物中Cl—Al键或P—Al键的键长,键级(MBO键级和FBO键级)和两单体之间的相互作用能。配合物中Cl—Al键或P—Al键的键长随着苯基的取代而不断减小,键级不断增大。配合物中单体之间的相互作用能分析结果:在A型配合物中,给电子的苯基增加了配合物中氯原子周围的电子密度,使氯原子上的静电势极小值由PCl3的-23.61 kJ·mol-1变到DPC的-90.56 kJ·mol-1,分子间相互作用能由-56.01 kJ·mol-1显著增加到-136.11 kJ·mol-1;在B型配合物的相互作用能也是随着苯基的加入而不断增大;配合物中单体分子之间的相互作用能越大,配合物分子就越稳定。
通过配合物单体之间的相互作用能研究,得出了类Friedel-Crafts酰基化制备BPD的反应过程中催化剂不能循环使用的根本原因:随着反应的不断进行,三种配合物中PCl3-AlCl3的相互作用能最小,化学活性最高,促进制备BPD的反应的顺利进行;其中BPD-AlCl3的相互作用能较小,有一定的化学活性,可以解离出催化剂AlCl3,能够使反应进行循环催化过程;DPC-AlCl3的相互作用能最大,解离出催化剂比较困难,导致催化剂不能循环催化反应,反应活性不断降低,这也是导致制备BPD的产率较低的原因。
单位:kJ·mol-1
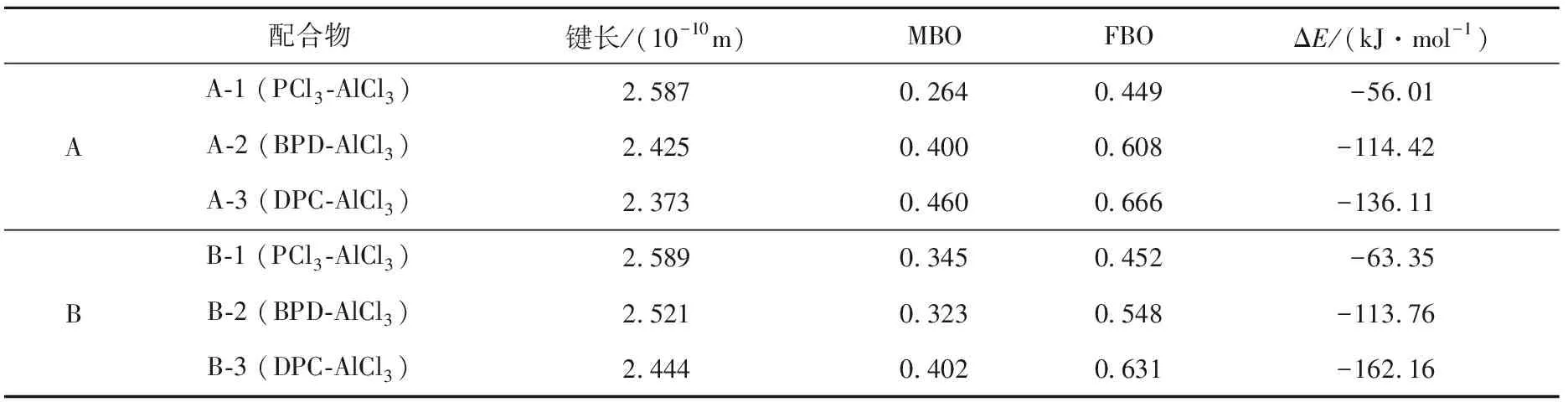
配合物键长/(10-10m)MBOFBOΔE/(kJ·mol-1)A-1 (PCl3-AlCl3)2.5870.2640.449-56.01AA-2 (BPD-AlCl3)2.4250.4000.608-114.42A-3 (DPC-AlCl3)2.3730.4600.666-136.11B-1 (PCl3-AlCl3)2.5890.3450.452-63.35BB-2 (BPD-AlCl3)2.5210.3230.548-113.76B-3 (DPC-AlCl3)2.4440.4020.631-162.16
2.3 QTAIM分析和LOL分析
为了更深入地了解配合物中单体分子之间成键的本质,对所有配合物分子进行了QTAIM[16]分析,其中配合物中的Cl-Al或P-Al原子之间的键临界点及键径证实了分子间成键作用的存在。表3给出了配合物中分子间相应键临界点处的电子密度拓扑分析数据。
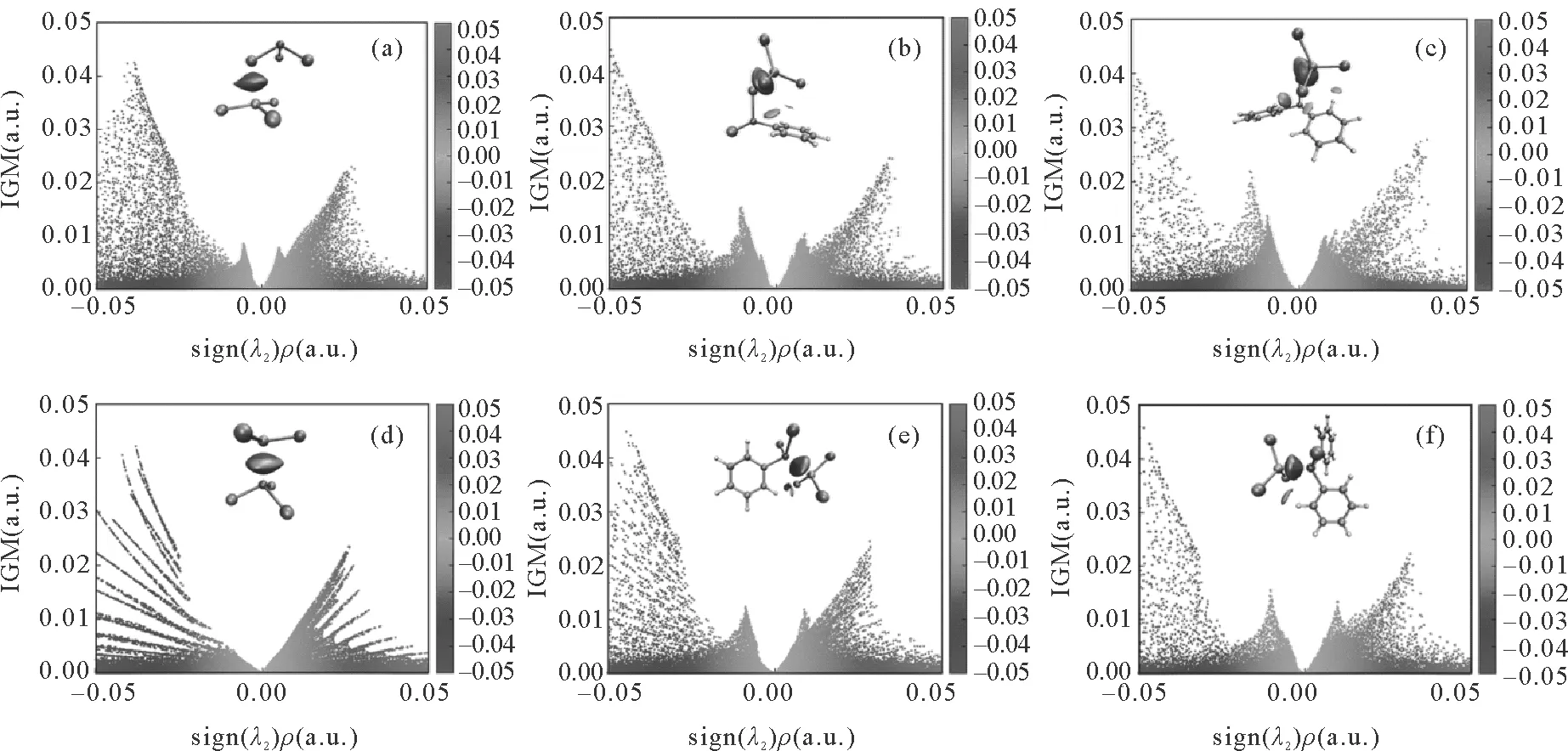
QTAIM理论指出,可以通过键临界点处的电子密度来研究化学键的强弱,通常键临界点处的电子密度ρ越大,则两个原子之间的化学键就越强,反之则越弱。Cl—Al键临界点上的电子密度ρ为0.024 8~0.038 9 a.u.,P—Al键临界点上的电子密度ρ为0.034 5~0.049 3 a.u.,相同的配合物中B型配合物的电子密度比A型的配合物的大。同一类型的配合物中键临界点处的电子密度ρ比较可得出:PCl3-AlCl3 表3 配合物中Cl—Al键和P—Al键的键临界点处的电子密度拓扑分析数据Tab. 3 Electron and energy densities at the bond critical points of complexes 定域化轨道指示函数LOL二维分布图可以清晰地看到原子周围的电子分布情况,可借此研究原子之间化学键的特征[18]。两种类型的配合物的Cl—Al键和P—Al键的二维LOL等值面图如图4所示,LOL清晰的展现了电子壳层的电子密度分布,通过观察在配合物中Cl—Al键和P—Al键的成键区域的电子密度分布情况,即可判断成键性质。 图4 配合物中Cl—Al和P—Al键径所在平面的LOL平面填色图Fig. 4 Two dimensional LOL maps of Cl—Al and P—Al bonds in complexes 由图4可知:A型配合物中氯原子最外层的电子密度有一些变形,箭头指示区区域偏向AlCl3中的Al原子,这说明单体与AlCl3形成了配位键,而且变形程度按PCl3-AlCl3 2017年,Corentin Lefebvre等[19]提出了一种可视化研究弱相互作用的方法-独立梯度模型IGM (independent gradient model),此方法受约化密度梯度函数RDG[20]的启发,但其基本原理不同,IGM给出了明确划分成片段间δginter和片段内δgintra两种数据(公式(1)和(2)),可以单独分析分子间或分子内的相互作用,而不受彼此的干扰。IGM方法给出的等值面图比RDG更为饱满一些,能更清晰的进行分子间弱相互作用的图示化展示。 (1) (2) 图5为配合物单体间相互作用δginter函数的平面散点图和IGM三维等值面图,其中等值面为δginter=0.01 a.u.。由散点图中的峰值分布可以得出,配合物中不仅存在配体与AlCl3中Al原子的强吸引作用(散点峰sign(λ2)ρ=0.05 a.u.附近),还有许多卤键,范德华作用力(散点峰-0.01 a.u. 图5 展示配合物单体间相互作用的IGM平面散点图和等值面图Fig. 5 IGM isosurface plots (isovalue=0.01 a.u.) are according to a BGR scheme via the interaction strengths 采用密度泛函理论对类Friedel-Crafts酰基化反应制备BPD过程中的PCl3-AlCl3、BPD-AlCl3和DPC-AlCl3配合物分子的理论分析表明:A型配合物中的PCl3-AlCl3和BPD-AlCl3形成能够使P原子上带有较正的静电势来进攻苯环,有助于反应的顺利进行;对于配体间的相互作用能对比分析,得出PCl3-AlCl3、BPD-AlCl3、DPC-AlCl3相互作用能依次增大;QTAIM及LOL图形化展示配合物中Cl—Al键或P—Al键成键特性,两种键都具有部分共价性质,而且B型配合物中的P—Al键的共价成分较A型配合物中的Cl—Al键的共价成分高;IGM图形化分析展示了配体之间存在较强的吸引力,卤键和少许的互斥效应,整体吸引作用克服了互斥作用,配体之间相互作用力强度的分析结果与计算的相互作用能的结果一致;由于配合物PCl3-AlCl3、BPD-AlCl3、DPC-AlCl3的相互作用强度依次增大,AlCl3从配合物中解离出来就越困难,一旦形成DPC-AlCl3就很难解离出AlCl3,直接导致催化剂不能进行循环催化。研究结果为未来芳烃与PCl3的实验研究中如何证实A型配合物和B型配位物的存在,如何避免形成DPC-AlCl3等问题提供了一定的理论参考。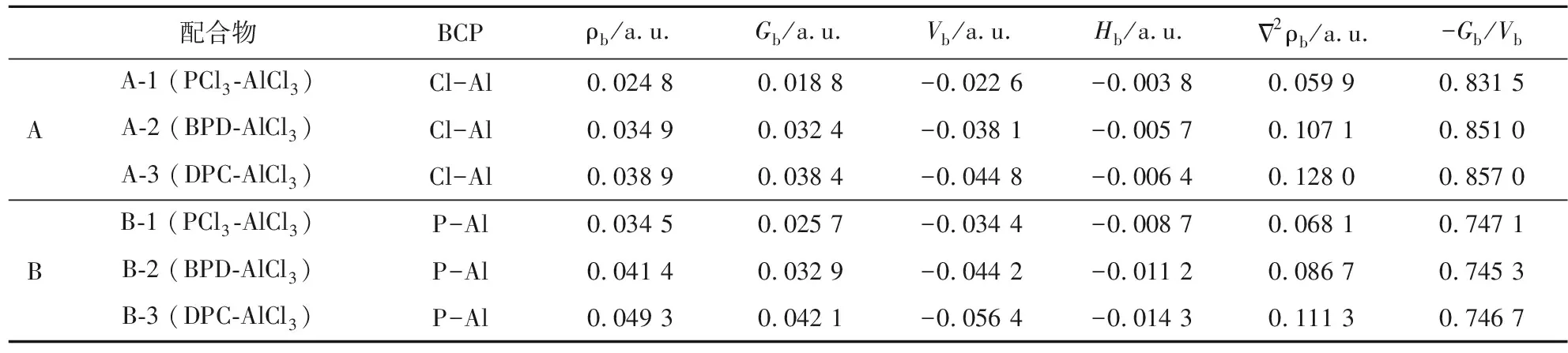
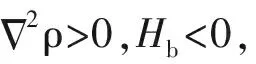
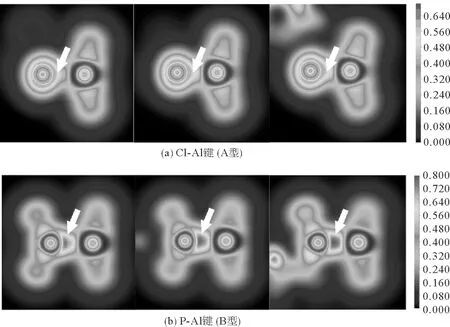
2.4 IGM函数等值面图形化研究相互作用
3 结论
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09测绘通报(2019年11期)2019-12-03赤峰学院学报·自然科学版(2019年5期)2019-09-10当代陕西(2019年6期)2019-04-17无机化学学报(2019年2期)2019-02-27科教导刊·电子版(2018年13期)2018-07-31中国洗涤用品工业(2017年2期)2017-04-16中国洗涤用品工业(2016年2期)2016-02-28烟草科技(2015年8期)2015-12-20科技与创新(2015年21期)2015-12-01
