
全基因组关联分析在乳酸菌研究中的应用
2019-11-13 01:23:06余中节宋宇琴孙志宏张和平
中国食品学报 2019年10期
余中节 赵 洁 宋宇琴 孙志宏 张和平
(内蒙古农业大学乳品生物技术与工程教育部重点实验室 农业部奶制品加工重点实验室内蒙古自治区乳品生物技术与工程重点实验室 呼和浩特 010018)
乳酸菌是指糖发酵能产生50%以上乳酸的一类无芽孢、不运动、过氧化氢酶反应阴性、革兰氏染色阳性细菌的总称[1]。乳酸菌是重要的工业用微生物之一,其能够使食品酸化,产生酒精,改善食品风味,维持肠道微生态,抑制病原菌生长,从而缓解乳糖不耐、腹泻、胃溃疡等病症,被广泛用于食品和生物医药行业。在青贮饲料发酵过程中,乳酸菌能够调节饲料内的生物区系,促进多糖与粗纤维转换,提高饲料转化率,促进反刍动物瘤胃发育;乳酸菌分泌的有机酸可以溶解土壤中的梨酸盐并通过螯合作用释放磷元素有助于植物生长。然而,传统的分析技术存在分辨率低,重复性差等缺点,未能全面解析乳酸菌的进化过程和遗传背景,在很大程度上限制了高生产性能和优良益生特性乳酸菌的应用领域和自身价值的发挥。
全基因组关联分析(Genome-wide association study,GWAS)的出现为乳酸菌的分离筛选以及溯源、进化研究提供了更为可靠的证据和新的思路。GWAS技术在细菌的基因-性状关联分析方面的应用已经渐趋成熟,这方面的研究也已成为细菌分子生物学的热门领域。本文综述GWAS在细菌中应用的现状,并分析了将GWAS分析技术应用于乳酸菌基因组研究及分离筛选的可行性,意在推动和倡导在我国加快对该项技术的应用研究。
1 乳酸菌分子生物学研究进展
人类对乳酸菌的应用有着长远的历史,根据《圣经·旧约》的记载,公元前 4000年,人类已经开始食用发酵肉制品及蔬菜腌渍物。1873年李斯特(Lister)利用稀释法,从酸乳中分离纯化出乳杆菌(Bacterium lactis),也就是目前的乳酸乳球菌(Lactococcus lactis),这是乳酸菌最早被分离出来的纪录[2]。目前在自然界发现的乳酸菌主要包括乳酸杆菌属(Lactobacillus)、乳球菌属(Lactococcus)、明串珠球菌属(Leuconostoc)、奇异菌属(Atopobium)等43个属373个种及亚种[3]。
据新思界产业研究发布的《2016-2019年中国乳酸菌市场深度分析报告》显示,在2015年我国乳酸菌的市场规模达到了660亿元,并且每年保持15%~18%以上的增速,而我国大部分乳酸菌市场被外资和合资企业所垄断,这是一种极不正常的现象,乳酸菌呈现出较强的地域性,特定地区的人群因其饮食习惯及肠道消化系统的差异,更适合本地区的乳酸菌,也就是习惯上所说的国民益生菌。然而,我国本土益生菌产品缺失,主要原因是对乳酸菌分离鉴定的基础研究和应用研究缺失,因而快速分离筛选出具有自主知识产权且生产、益生性能良好的乳酸菌,以及阐明乳酸菌的分离学地位、进化过程和遗传背景,对发展民族乳酸菌产业非常重要。
早期对于乳酸菌的研究主要集中在利用其生理生化特性进行分类鉴定,以及分离筛选,这种手段一是要通过大量的实验得出结论,时间成本和实验成本很高;二是生物的表型与其基因型并不完全对应,因而该方法存在分辨率低,工作量大等诸多限制。分子生物学的发展使得乳酸菌的快速分离筛选,以及阐明乳酸菌的起源、进化成为可能。由此产生的一系列新型技术,如以16S rRNA基因间隔区(Intergenic spacer region,ISR)序列分析技术为代表的DNA指纹图谱技术,为乳酸菌的分离鉴定提供了丰富的方法。
基因芯片技术是上世纪90年代兴起的一种对成百上千甚至上万个基因同时进行检测的新技术,有高通量、并行化等特点。Frese等[4]利用比较基因组杂交(Comparative genomic hybridization,CGH)技术对57株分离自不同脊椎动物的罗伊氏乳杆菌(Lactobacillus reuteri)的基因组多态性进行研究,发现分离自啮齿动物的菌株在聚类分析中形成了单独的分支,表明其与其它5个分离源的分离株在基因组上存在明显差异。当然该方法也存在一定的局限性,首先其只能将参考基因组中的有限基因用于设计基因阵列,其次低杂交率的基因将不能用于研究[5]。
随着逐步克隆法和全基因组鸟枪法等一代测序技术的出现,对乳酸菌基因组的研究进入了测序时代。2014—2016年内蒙古农业大学教育部重点实验室“乳品生物技术与工程实验室”采用高分辨率的多位点序列分型技术(Multilocus sequence typing,MLST)对包括分离自中国、蒙古国和俄罗斯等地的多个乳酸菌进行研究[6-13],阐明菌株基因重组与变异对其进化的贡献,进化与世系、分离地和分离源的关系,加深了对乳酸菌遗传多样性和群落结构的理解,并对菌株分类学地位的划分提供新的建议,为乳酸菌的溯源提供了新的理论依据。然而,在试验研究过程中,由于生物繁殖中不可避免的同源重组现象,使得菌株间垂直遗传信号受到干扰,因此仅依靠几个靶标基因序列无法还原菌株真实的垂直进化关系[14];在菌株分离筛选层面,MLST技术也只能进行推测,Song等[12]利用将与工业用菌株位于同一世系的分离株为依据来推测可能用于工业生产的新菌株,缩小了人工筛选的范围,却未能给出具体影响生产性能的基因。
之后随着计算和生物技术的不断发展,测序成本快速降低,通量不断提高,对乳酸菌基因组的大规模测序得以实现。2001年Bolotin等[15]完成对第一株乳酸菌-乳酸乳球菌乳酸亚种(Lactococcus lactis ssp.lactis)IL1403的全基因组测序。2010年,Zhang等[16]完成了我国第一株乳酸菌——干酪乳杆菌 Zhang(Lactobacillus casei Zhang)的全基因组测序。Sun等[17]、Zhong等[18]分别对45株双歧杆菌(Bifidobacterium)和37株肠球菌(Enterococcus)进行了比较基因组学分析,找到了双歧杆菌属和肠球菌属的祖先,阐明了双歧杆菌和肠球菌的传播过程。2015年,Sun等[19]完成了对213株乳杆菌(Lactobacilli)的重测序,并重构了系统发育树,建议将乳杆菌属、片球菌属(Pediococcus)、魏斯氏菌属(Weissella)、明串珠菌属(Leuconostoc)、嗜果糖乳酸菌属(Fructobacillus)和酒球菌属(Oenococcus)命名为乳杆菌属复合体(Lactobacilli complex),并详细阐述了蛋白代谢和糖代谢的基因,同时解析了CRISPR-Cas免疫系统,为将来对乳杆菌的基因编辑打下了基础。全基因组序列虽然能够更完整的表达乳酸菌的进化过程,但其也仅能将基因存在/缺失的数据利用Phenolink等[20]、CFAS等[21]相关性分析工具找到具体某个基因的存在与缺失对特定表型的影响,忽略了基因结构、拷贝数、基因间隔区、基因表达能力等差异对表型的影响。GWAS是在全基因组层面上开展大样本、多中心、重复验证的技术,并对相关基因与复杂性状进行关联研究,从而能够全面揭示出乳酸菌生长速度、产酸量、耐高温、耐酸、耐胆盐等性状的遗传机制,同时也为乳酸菌的分离筛选提供更可靠的证据。
2 全基因组关联分析(Genome-wide association study,GWAS)
从1865年Gregor Johann Mendel“颗粒遗传”概念的提出到1953年WatsonCrick提出DNA分子的双螺旋结构模型;从20世纪70年代Fred Sanger及其同事发明第一代DNA测序技术到2005年以 Solexa、ABI SOLiD为代表的第二代DNA测序技术的研发与应用,从1977年Fred Sanger完成对全长为5 375 bp的PhiX174噬菌体基因组的测定到2003年人基因组计划(Human genome project,HGP)的完成,人类完成了对遗传物质从认识到结构解析再到精准测序的演变。随着人类基因组计划的完成、基因芯片和第二代基因组测序技术的大规模应用,标志着人类正式进入后基因组时代,全基因组关联分析应运而生。
2.1 GWAS的概述
全基因组关联分析最初应用于人类基因组,其原理是应用基因组中数以万计的单核苷酸多态性(Single nucleotide phlymophism,SNP)、插入和缺失(Insertions and deletions,InDels)或拷贝数变异(Copy number variations,CNV)进行病例-对照的关联分析,以发现影响疾病发生的遗传特征[22],之后随着高通量测序技术的发展,也逐渐应用于其它模式和非模式的动植物中,甚至是一些经典的原核生物中。第一篇在人类中成功应用GWAS的文献于2005年在Science上发表[23]。Klein等[24]在96个老年性黄斑病变的病例和50个同样来自非西班牙裔的白人作为对照组的研究中共找到116 204个SNPs位点,发现其中位于补体因子H的内含子中的SNP rs3380390和rs1329428与疾病显著相关。虽然对于GWAS方法的某些方面仍然存在争议,但截止到2013年National human genome research institute(NHGRI)已经有 1 751篇GWAS文章被出版,共找到11 912个SNPs与疾病相关[25]。
2.2 GWAS的原理及分析流程
GWAS分析的主要原理是在群体中选择病例组和对照组或连续分布的数量性状,比较全基因组范围内所有遗传标记位点的等位基因或基因型频率在病例组和对照组之间的差异,若某个遗传标记位点的等位基因或基因型在病例中出现的频率明显高于或低于对照组,则认为该位点与性状存在关联,之后根据该位点在基因组中的位置和连锁不平衡关系推测与性状相关的基因。全基因组关联分析的主要流程包括样品采集、表型数据的测量、全基因组测序、基因组拼接、SNP、idnel、CNV等分子标记检测、群体遗传分析、连锁不平衡分析、性状关联分析和目标性状相关区域基因功能注释。
2.3 GWAS的优势
相关性分析总体上可以分为“自下而上”和“自上而下”两种方法。“自下而上”,即以基因组为基础方法,以DNA序列为基础,检测其对表型的影响,该方法已被广泛应用,然而存在诸多限制,例如:其工作量较大,无法应用在共生细菌中,只能判断基因的存在与缺失对表型的影响,而无法判断基因的调控或蛋白质的多态性对表型的影响。“自上而下”,即从表型开始,检测特定的基因组区域与表型的不同联系,也就是GWAS[26]。GWAS是以自然群体为研究对象,以长期重组后保留的基因间的连锁不平衡(Linkage disequilibrium,LD)为基础,将目标性状表型的多态性与基因的多态性相结合,共同分析,能够直接鉴定出与表型密切相关且具有特定功能的基因位点,扩大了相关性分析的应用范围,同时实现了在提高相关性信号分辨率的基础上减小试验工作量。
2.4 GWAS的应用条件
无论将GWAS方法应用在人类或是细菌中,其都必须满足以下3个先决条件,即可测的表型和可测的基因型,以及足够样品数量来保证统计学效力,并在此前提下选择正确的统计分析方法。众所周知,更多样品的统计学可靠性更好,所以为了保证基因组的异质性以及数据的正态性,应保证样品数不少于30个[27]。
2.5 GWAS在细菌中的应用现状
GWAS是基因组分析的重要工具,并且近几年已经成功应用到细菌基因组分析上(表1)。早期的细菌基因组主要基于PCR技术或比较基因组杂交技术,通过与表型结合,以寻找基因组中特定基因的存在或缺失与表型的关系,然而因为成本和平台的限制,以及某些细菌自身的核苷酸多样性较低,如炭疽杆菌(Bacillus anthracis)[28],使得其无法在细菌基因组中大规模应用。2013年Sheppard等[29]首次实现GWAS在细菌中的应用,将192株分离自牛和鸡肠道的弯曲杆菌(Campylobacter)的DNA序列划分成30 bp长的“word”来寻找菌株与特定宿主定殖能力方面的相关性,共检测到7 307个“word”与弯曲杆菌在牛肠道中的特异性定殖有关,注释到3个与维生素B5生物合成相关基因,因而认为弯曲杆菌自身能否合成维生素B5是其能否在牛肠道定殖的关键。之后Chewapreecha等[30]的研究首次使GWAS在细菌中的研究达到了人类遗传学研究的规模,他们总共分析了收集自缅甸和泰国边境的3 085株和收集自美国马萨诸塞州的616株肺炎链球菌(Streptococcus pneumoniae)用于检测β-内酰胺抗性相关的基因组变异。
结核分枝杆菌(Mycobacterium tuberculosis),俗称结核杆菌,是引起结核病的病原菌。Farhat等[31]利用基于进化收敛性的方法找到123株结合分枝杆菌的独立突变位点,并以卡内蒂分枝杆菌(Mycobacterium canettii)作为外群构建系统进化树,验证了全部前人研究发现的抗性基因并且发现39个主要编码细胞壁生物合成、转录调控和DNA修复通路的基因与分枝结核杆菌的抗性有关。D-环丝氨酸是治疗结核病的高毒性二线药物,Christopher等[32]通过对498株结核分枝杆菌的相关性分析,找到21个基因的功能缺失突变与常用于治疗结核病9种药物的耐药性有关,其中编码L-丙氨酸脱氢酶的ald基因拥有11种不同的功能缺失突变与D-环丝氨酸的抗药性有关,通过基因敲除试验验证了相关性分析结果。
金黄色葡萄球菌(Staphylococcus aureus,S.aureus)是人体皮肤和鼻腔的常见定殖菌。Laabei等[33]通过对收集的90株耐甲氧西林金黄色葡萄球 菌(Methicillin-resistant S.aureus,MRSA)的GWAS分析,发现了121个基因座位与分离株的不同毒性有关。同样,Alam等[34]在49株万古霉素敏感金黄色葡萄球菌(Vancomycin-sensitive S.aureus)和26株万古霉素中度敏感金黄色葡萄球菌(Vancomycin-intermediate S.aureus)中检测到55 977个SNPs,发现编码RNA聚合酶的rpoB基因第481位密码子的非同义替换与金黄色葡萄球菌的万古霉素抗性显著相关。
由上述研究可知,GWAS已经有在细菌中成功应用的先例,成功的定位弯曲杆菌、肺炎链球菌、结核分枝杆菌和金黄色葡萄球菌等细菌的毒力基因和抗性基因,因而GWAS技术在定位乳酸产酸量、产酸速度等与生产特性相关的基因,以及阐明分离源、分离地等选择压力对乳酸菌进化的影响充满了希望,然而同时也面临着挑战。
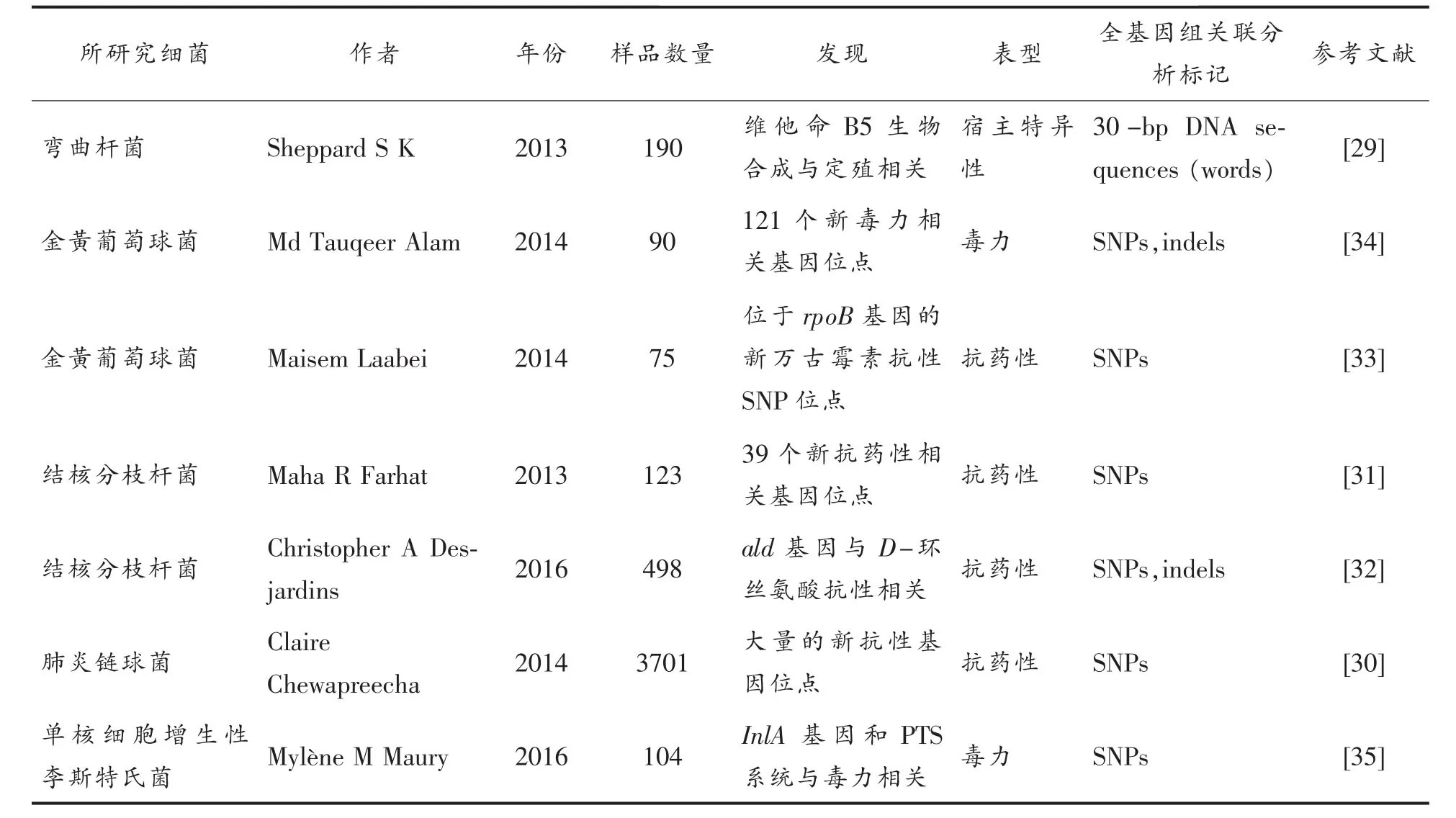
表1 近几年全基因组关联分析在细菌中应用的文献Table1 Genome-wide association study application in bacteria in recent years
2.6 GWAS在细菌基因组中应用存在的限制
2005年二代测序技术的出现使得对细菌基因组的大规模测序得以实现,而GWAS并未立即应用于细菌基因组分析。究其原因,主要是由于原核细胞与真核细胞在组成上存在差异,细菌是单倍体生物,缺乏着丝粒与纺锤体结构,双链环形DNA随着细胞的伸长采用二分裂的方式分开,因而使其无法像真核细胞在减数分裂四分体时期非姐妹染色单体之间发生同源重组,从而导致细菌基因组出现严重的种群分层和较高的连锁不平衡现象,也就使得在人类基因组中广泛应用的GWAS分析无法直接套用于细菌基因组中。
2.7 GWAS在细菌中应用所面临困难的解决方法
群体分层的问题不仅在细菌的GWAS研究中出现,同样也出现在人类的GWAS研究中,即在病例对照的研究中,如果某遗传标记的等位基因的频率在病例和对照组中出现显著差异,而该遗传标记并不与表型相关,则认为该研究中存在群体分层现象[35],简单来讲就是等位基因在亚群中非随机分布。因为细菌特殊的克隆繁殖方式使得其群落高度相关,导致细菌的GWAS研究受到群体分层的严重干扰,可是这往往被研究者所忽视,相关性结果的出现是因为遗传的相关性而不是基因组的变异,从而使得相关性检验的P值膨胀,与表型相关的和不相关的突变同时关联到同一个连锁区域内,从而出现假阳性的结果,使得研究结果难以重复。解决群体分层的方法可以通过有选择性的采样,采集隔离或来自同一家系的不同表型的群体样品,Maury等[36]通过该方法收集了104株单核细胞增生性李斯特氏菌(Listeria monocytogenes),通过GWAS分析发现内化蛋白基因InlA的截短是低毒性李斯特菌的主要变异特征,而一个由6个基因构成的磷酸转移酶系统(Phosphotransferase system,PTS)是高毒性李斯特菌的主要毒力因子。另外,可以通过使用线性混合模型(Linear mixed model,LMM)来消除不同祖先或有相关性的样品造成的有偏倚的相关性。Earle等[37]使用该方法用于查找来自4个不同种的3 363株致病菌在17种药物中的潜在的抗性基因。还可通过使用重建系统发育树[31,34],或聚类后进行大量置换检验的方法[33]以及基因组控制[38]等方法来消除。
连锁不平衡是指一个基因座位上的某些等位基因与同一个染色体上另一个基因座位上的某些等位基因同时出现的频率大于单独随机出现的频率,连锁不平衡的程度可以用r2来衡量[39]。同样由于细菌无性克隆的繁殖方式以及单倍体基因组结构,使得其只能通过接合、转化和转导产生重组和交换遗传信息,造成细菌基因组的高度连锁不平衡,通过连锁区域中其它连锁位点使真正与特定表型有因果关系的变异位点变得模糊,从而限制经典的基因组作图工具,只能将有相关性的区域定位到较长连锁片段,形成所谓的“马赛克基因”,降低了相关性信号的分辨率。要解决连锁不平衡最直接的办法就加大样本量,Chewapreecha等[30]使用了3 701株肺炎链球菌,为GWAS分析提供了足够的重组事件。减少连锁不平衡的不利影响第一步是要明确所研究细菌连锁不平衡的水平,four-gamete test[40]或 D′measure[41]等工具可以评价细菌的连锁不平衡水平,之后可以通过定义一个变异是否能够引起表型功能上的改变,例如抗药性、菌落总数等,来确定该变异是否为因连锁不平衡造成的假阳性变异[42]。同时还可以结合细菌基因组具有显著的正向选择的特点,来提高GWAS的分辨率。
3 GWAS在乳酸菌中应用的意义
GWAS利用广泛分布于基因组中的SNP,In-Del,CNV等分子标记,将目标性状表型的多态性与基因的多态性相结合,共同分析,相比于MLST技术、比较基因组学技术具有更高的分辨率,能够在单核苷酸的水平解析乳酸菌生产性能的不同,为乳酸菌的分离筛选提供更可靠的证据。在遗传进化方面,不仅可以验证前人的研究结果,还能够提供更多、更完善的信息,为乳酸菌的溯源、遗传多样性的研究提供新的思路。目前GWAS在细菌基因组中的应用尚处于发展初期,而且已完成的研究也主要集中在致病菌中,在包括乳酸菌等工业用微生物中还鲜有报道[43]。目前我研究团队正在开展对于嗜热链球菌(Streptococcus thermophilus)的GWAS研究,在已完成的研究中发现,以分离源和发酵特性分别做为表型,发现冷激蛋白(Cold-shock protein)和钙转移 P型 ATP酶(Calcium-translocating P-type ATPase)、胞壁质水解酶调控酶lrgA(Murein hydrolase regulator LrgA)、膜蛋白(Membrane protein)分别与菌株的分离地的海拔和产酸量、产酸速率呈显著相关关系。早期研究已经证明GWAS不仅是一个寻找特定表型与其所关联的基因型的可靠方法,而且是一个揭示复杂性状表达水平的强有力的工具[44],因而将GWAS应用于乳酸菌等工业微生物的分离筛选、进化以及溯源将具有开创性意义。
人类对于乳酸菌的应用有着很长的历史,对于乳酸菌的研究已经深入到分子水平,GWAS技术可以使乳酸菌的基因组研究精确到核苷酸水平,为乳酸菌的分离筛选以及进化溯源提供更可靠度证据和新的思路。我国立足于民族乳酸菌从收集、筛选、分离、鉴定等基础和应用研究起步较晚,且与发达国家存在较大差距,而GWAS技术的应用为我们在较短时间内缩短与发达国家的差距提供了可行的途径。为此,将GWAS技术应用于乳酸菌的遗传和进化研究以及分离筛选,具有更加现实的意义。
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13 06:38:44
今日农业(2021年11期)2021-08-13 08:53:24
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国洗涤用品工业(2015年7期)2015-02-28 19:02:39
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
中国酿造(2014年9期)2014-03-11 20:21:04
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
