
间[*I]苄胍的合成
2019-09-03 11:31陈志明吴二明黄荷云
药学与临床研究 2019年4期
陈志明,王 刚,汪 洋,吴二明,黄荷云
国家卫生健康委员会核医学重点实验室,江苏省分子核医学重点实验室,江苏省原子医学研究所,无锡 214063
间碘苄胍 (Metaiodobenzylguanidine,MIBG)是去甲肾上腺素神经递质(NE)的一种同系物,借助于NE,可以被富含交感神经的细胞所吸收。它是一种在诊断和治疗心肌异常及神经内分泌瘤方面非常宝贵的载体,125I-MIBG可用于PET显像,123I-MIBG可用于心肌显像,131I-MIBG也被广泛用于神经内分泌瘤的显像和治疗。目前临床上使用的[*I]MIBG是采用同位素交换法制备的,这种方法制得的药液中含有大量的未标记的MIBG载体,而这些未标记的MIBG在给药过程中会与标记的 [*I]MIBG竞争进入目标细胞,降低细胞对有效成分的吸收。如果能把未标记的MIBG完全去掉——即制备无载体的MIBG,则其药效可能会大大提升[1-3]。
为了提高药效和降低可能的副作用,无载体[*I]MIBG的合成方法被广泛研究。Hunter DH等[4]报道了利用固相合成的方法在树脂上制备无载体[*I]MIBG(合成路线见图1),但此方法合成复杂,树脂不易获取。
Ganesan V等[5,6]报道了分别以间位三甲基硅基苄胍和N,N-二叔丁氧羰基,3-三丁基锡基苄胍为前体合成无载体[*I]MIBG的方法,前者合成步骤繁多,标记复杂;后者在前体中引入的BOC保护基团,在标记的时候会以杂质的形式残留在体系中,影响后续药效研究。合成路线见图2。
本课题组在合成含BOC保护基团的无载体[*I]MIBG锡前体的基础上[7],再次开发出以间碘苄胍的碳酸氢盐为原料,一步得到目标化合物3-三丁基锡基苄胍作为不含保护基团的无载体 [*I]MIBG锡前体。此合成反应条件温和,步骤简便,收率较高,适合实验室制备,而且在标记的时候不会引入多余的杂质基团,利于后续药盒的研发。

图1 Hunter DH等报道的合成路线图

图2 Ganesan V等报道的合成路线图
现对得到的锡前体进行了初步的标记研究。见图3。
1 材 料
1.1 仪器

图3 本研究的合成路线图
TENSOR-27 FT-IR型红外光谱仪、AvanceⅢ400核磁共振谱仪 (德国Bruker公司);SQ Detector 2质谱仪 (美国 Waters公司);Calirad Isotope Calibrator CRC-250放射性活度计 (美国Victoreen公司);γ-计数器(γ-counter 2480,美国 PE 公司);Waters 1525型高效液相色谱系统 (1525高压泵和2487紫外检测器),Waters Symmetr C18(4.6mm ×150mm,5μm)色谱柱。
1.2 药品与试剂
间碘苄胺盐酸盐(上海毕得有限公司);碘[131I]化钠溶液(批号 20170108,规格 7.51GBq·mL-1,放射化学纯度98.6%,成都中核高通同位素有限公司);氨基腈(NH3CN)、双(三丁基锡)((SnBu3)2)、双三苯基磷二氯化钯[Pd(PPh3)2Cl2]、氯胺 T、双氧水(均为Sigma Aldrich);其余试剂均为化学纯。
2 方法和结果
2.1 化合物1
化合物1由间碘苄胺盐酸盐为原料,参照文献方法合成得到[8]。m.p.124℃~126℃(文献 124℃~126 ℃)。IR (KBr)ν(cm-1):1628,1696,3061,3472。1H-NMR (500 MHz,CDCl3) δ4.34 (-CH2,s,2H),7.10~7.14 (Ph-5,t,1H),7.34~7.36 (Ph-6,d,1H),7.63~7.65(Ph-4,d,1H),7.69(Ph-2,s,1H);ESI-MS[M+H]+:276。
2.2 3-(三丁基锡基)-苄胍(2)的合成
在50mL的圆底烧瓶中,加入500mg(1.48mmol)化合物1溶于10mL二甲亚砜(DMSO),再加入947mg(1.63mmol)双(三丁基锡)溶于 30mL 1,4-二氧六环,最后加入104mg(0.15mmol)双三苯基磷二氯化钯,在100℃下恒温反应至溶液变黑。离心旋干,用乙酸乙酯溶解后水洗3遍,离心再旋干,甲醇溶解后用正己烷洗3遍,将多余的锡试剂洗掉,后加无水硫酸钠干燥。用制备色谱分离得453mg淡黄色液体,收率 68.7%。1H-NMR(400MHz,CD3OD) δ0.88~0.91(Bu-CH3,m,9H),1.08~1.11(Bu-CH2,m,6H),1.32~1.37(Bu-CH2,m,6H),1.55~1.58(Bu-CH2,m,6H),4.39 (-CH2,s,2H),7.24~7.26 (Ph-5,m,1H),7.33~7.37(Ph-6,m,1H),7.41(Ph-4,Ph-2,m,2H);ESI-MS[M+H]+:439.4。
2.3 3-(三丁基锡基)-苄胍(2)的标记
在 0.25mL(约 0.5mCi)[131I]碘化钠溶液中加入50μg化合物 2,0.5mL 磷酸二氢钾(pH=6.5)缓冲溶液(按《中国药典》2015版方法配制),10μL氯胺T溶液(50mg·mL-1)后振荡,常温反应 5min,再加入10 μL Na2S2O5溶液(100mg·mL-1)终止反应,HPLC法分析结果。
图4是化合物2标记后溶液紫外检测的HPLC图谱;图5是放射性检测γ计数的HPLC图谱。在RT=5min处的吸收峰对应的是 [131I]-MIBG的吸收峰,与纯的MIBG在同一系统中的保留时间吻合,以峰面积计算[131I]-MIBG标记率达到89.4%。

图4 化合物2标记后的紫外检测HPLC图谱

图5 化合物2标记后的放射性HPLC图谱
3 讨 论
在化合物2中,由于强极性的胍基团的存在,使得无法用常规的柱层析来分离提纯,这对反应的分离纯化提出了挑战。Ganesan V等[5,6]为了解决这个问题,在化合物中引入了Boc保护基团,降低了分子的极性,使得能用常规的柱层析来分离;但是这样做的缺点也显而易见,在后续的标记过程中,Boc保护基团将会以杂质小分子的形式残留在药液中,可能对后续药液的纯度产生影响。
这在当时也许只能用这种间接的方法来分离提纯含胍基的化合物,但是随着制备色谱的广泛应用,使得在实验室中大量分离提纯含胍基的这类强极性的化合物成为可能,所以本研究果断放弃了在分子中引进其他辅助基团的做法,而是直接用制备色谱分离提纯目标产物。
需要指出的是,特意选用间碘苄胍的碳酸氢盐作为起始原料,而不是经典的硫酸盐,是因为目标化合物含锡基团,在酸性条件下易离去,被氢离子取代,不利于反应产物的稳定。
在对化合物2进行标记研究的时候,选择了常用的两组氧化剂进行比较对照——氯胺T和双氧水。固定化合物 2(50μg)和131I溶液(0.5mCi)的量,分别考察了不同量的氧化剂对标记率的影响,即得到图6的结果。
由图6看出,大体上氯胺T的量越多,化合物2的标记率越低,这可能是因为随着氧化剂量的增多,其氧化能力越来越强,导致氧化过程产生的副产物增多。以双氧水作为氧化剂时也观察到了相似的现象。另外,值得关注的是,同样多量的氧化剂,双氧水的标记率要超过氯胺T,这同样可能与氯胺T的氧化性强于双氧水有关。双氧水的标记率峰值在93%左右,而氯胺T的峰值只有89%。
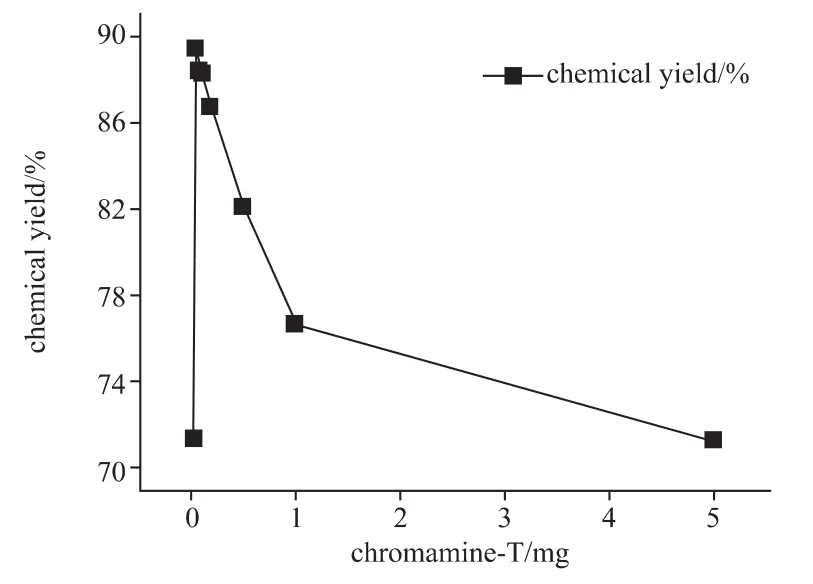
图6 氯胺T的量对最终放射化学产率的影响曲线
同样,对标记的反应时间也进行了考察,氯胺T标记达到峰值所需的时间大约是5min,而双氧水标记达到峰值所需的时间则大约是2 h。
4 结 论
本研究从简单原料间碘苄胍的碳酸氢盐开始,通过一步反应制得了无载体MIBG的锡前体,可以直接用于制备无载体MIBG,且标记率在85%以上。该合成方法简便易行,条件温和,适合在大多数实验室合成,而且得到的产物在标记的时候无多余杂质,适合用于后续药盒的研究。
致谢
感谢江南大学食品工程学院提供的核磁和质谱测试。
猜你喜欢
太原理工大学学报(2022年2期)2022-03-21
化工环保(2021年3期)2021-06-17
染整技术(2020年5期)2020-06-04
合成技术及应用(2020年1期)2020-01-12
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
浙江农业学报(2017年1期)2017-05-17
中国资源综合利用(2016年6期)2016-01-22
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中学生数理化·高二版(2008年5期)2008-11-12
