
祁连山中段退化高寒草地土壤细菌群落分布特征
2019-08-30 02:27:00李海云姚拓马亚春张慧荣路晓雯杨晓蕾夏东慧张建贵高亚敏
草业学报 2019年8期
李海云,姚拓,马亚春,张慧荣,路晓雯,杨晓蕾,夏东慧,张建贵,高亚敏
(甘肃农业大学草业学院,甘肃 兰州 730070)
草地在陆地生态系统中起着重要作用,尤其是在营养物质和能量循环过程扮演着重要角色[1]。近年来由于对草地资源的过度放牧和滥耕乱伐,使草地植被破坏严重,水土流失加剧,草原鼠虫害严重[2-3],导致草地退化、沙化、盐碱化面积日益扩大,近年来草地退化等生态问题已受到人们的普遍关注和高度重视[4]。放牧活动是影响高寒草地退化的主要驱动因素之一,由于家畜的采食、践踏和排泄粪便等改变了地表植被覆盖状况,造成地表特征的改变,进而影响土壤结构的破坏、养分的损耗、土壤微生物区系和数量的改变,最终导致草地退化[5]。随着研究和实践的深入,许多研究者认识到草地退化不仅是地表植被和土壤理化特性的退化,更重要的是草地土壤微生物群落结构和多样性的变化。土壤微生物作为土壤生态系统的重要组成部分,在土壤有机质分解和营养物质循环等生态系统功能过程中起主导作用[6]。由于土壤微生物对生存的微环境十分敏感,能对土壤环境变化做出快速反应,进而影响植物群落物种多样性和土壤结构的形成。因此,土壤微生物群落结构和多样性的变化可作为衡量草地生态系统的健康变化和草地退化或恢复程度的重要指标[7]。目前,关于草地退化对土壤微生物的研究,主要集中在可培养微生物、磷脂脂肪酸(phospholipid fatty acid,PLFA)、PCR-DGGE、16S rRNA基因文库构建技术等方面[8-11]。这些传统的研究方法所能反映出的土壤微生物信息十分有限,在很大程度上低估了土壤微生物的物种组成并高估其丰度[12]。随着高通量测序技术的兴起,其具有高准确性、高灵敏度等优势,能够较为全面和准确地反映土壤微生物群落结构,被广泛应用于各研究领域中[13],弥补了前期土壤微生物传统研究方法的不足。因此,为探究祁连山中段不同退化草地土壤细菌群落结构变化规律,采用高通量测序技术分析退化高寒草地土壤微生物群落变化趋势,为高寒草地退化与恢复治理提供理论依据和数据参考价值。
1 材料与方法
1.1 研究区概况
研究区位于祁连山中部地段(37°53′ N、101°45′ E),隶属于甘肃省张掖市肃南县皇城镇,其南部为冷龙岭山地,北部为盖掌达坂山地,东连天祝藏族自治县,西接山丹军马场。海拔2600~3500 m,属高原大陆性气候,全年平均气温0.6~3.8 ℃,无霜期不足140 d,绝对无霜期仅45~60 d,年降水量361.6 mm,气候寒冷多变,四季不分明[14]。
1.2 样地设置及土样采集
依据草地退化程度划分相关划价标准[15],在2017年8月对肃南县皇城草地植被进行调查,选择3种退化草地类型,分别为:轻度退化草地(lightly degraded grassland,LDG)、中度退化草地(moderately degraded grassland,MDG)和重度退化草地(severely degraded grassland,SDG),具体样地信息见表1。在每种样地中,设置3个大样方(50 m×50 m),其内随机设置3个小样方(50 cm×50 cm),记录小样方中植物种类、物种数、盖度和高度。地上部生物量采用齐地面刈割,将鲜样在65 ℃下烘干24 h后称重。用土钻(内径53 mm)采集0~20 cm土壤样品,土样混匀后装入已灭菌袋中迅速带回,在-80 ℃冰箱中保存以备用。土样分为两份,一份用于土壤养分及酶活测定,另一份用于土壤微生物总DNA提取。

表1 样地基本信息Table 1 Basic information of the soil samples
1.3 土壤理化性质及酶活性测定
采用鲍士旦[16]的方法测定土壤理化性质pH、含水量、电导率、有机质、全量养分和速效养分含量;采用关松荫[17]的方法测定土壤脱氢酶、氧化还原酶、脲酶、中性磷酸酶、纤维素酶和蔗糖酶活性。
1.4 土壤总DNA提取及细菌16S rRNA基因扩增
采用OMEGA土壤DNA提取试剂盒(上海翊圣生物科技有限公司,上海)对土壤DNA基因组进行提取,每个样品3次重复并进行混合,以降低DNA提取过程中造成的误差。以提取的DNA为模板(1 ng·μL-1),对细菌16S rRNA基因V4~V5区采用带Barcode的特异性引物[18](515F和907R)进行PCR扩增,每个样品3次重复。PCR扩增条件参照赵帆等[19]的方法进行扩增。PCR产物根据产物浓度进行等量混样,充分混匀后使用2%的琼脂糖凝胶电泳检测PCR产物并切胶回收,产物送至天津诺禾致源生物信息科技有限公司,采用Illumina HiSeq PE250高通量测序技术平台进行测序分析。
1.5 生物信息学分析
根据Barcode序列和PCR扩增引物序列从测定数据中拆分出各样品数据,截去Barcode和引物序列后对每个样品的reads进行拼接[20]后得到原始测序数据;原始测序数据经质检和嵌合体去除得到有效数据(effective tags)。利用Uparse软件对样品的有效序列进行OTUs聚类(相似度97%以上),用Mothur方法[21]与SILVA软件的SSUrRNA数据库[22]进行物种注释(阈值:0.8~1.0);采用PyNAST软件[23]与GreenGene数据库中数据信息进行多序列比对,最后对测序数据进行标准化处理,采用Qiime 1.7.0软件计算Alpha多样性指数和Beta多样性。采用R 2.15.3软件的Vegan软件包绘制稀释曲线,并进行无度量多维标定法分析(non-metric multi-dimensional scaling,NMDS)。
1.6 数据分析
所有数据均采用SPSS 21.0软件进行处理分析,One-Way ANOVA和Duncan氏新复极差法分析差异显著性。采用CANOCO 4.5软件对细菌优势群落进行去趋势对应分析(DCA),根据第一排序轴的梯度范围(lengths of gradient)数值,小于3.0,采用冗余分析(RDA);为3.0~4.0,采用RDA分析和典型相关分析(CCA)均可;大于4.0,采用CCA分析,对细菌群落与土壤环境指标间的相互关系进行分析。
2 结果与分析
2.1 植被特征
3种高寒草地植被特征间差异显著(P<0.05)。随着退化程度的加重,植被盖度、高度、地上生物量均明显降低,依次为轻度退化>中度退化>重度退化(表2);群落物种多样性和均匀度指数的变化为轻度退化>中度退化>重度退化。
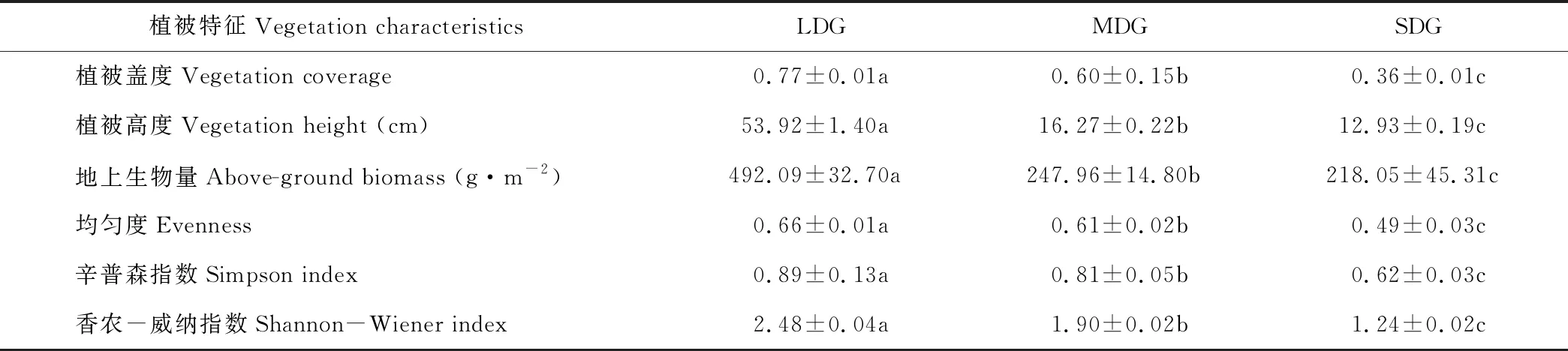
表2 各样地植被特征Table 2 The basic vegetation characteristics of different soil sampling sites
注:同行不同字母表示差异显著(P<0.05),下同。
Note: Different letters in the same line mean significant differences at 0.05 level, the same below.
2.2 土壤理化性质
从表3可以看出,各样地土壤理化性质及酶活性间均差异显著(P<0.05)。随着草地退化程度的加重,土壤含水量、有机碳、有机质、全氮、全磷、全钾、速效磷、速效氮、速效钾均呈逐渐减小趋势;土壤电导率呈逐渐增大趋势;土壤pH、脲酶、中性磷酸酶、脱氢酶、蔗糖酶和氧化还原酶均呈先增大后减小趋势;土壤纤维素酶呈先减小后增大趋势。
2.3 土壤细菌群落丰度与Alpha多样性分析
通过Illumina HiSeq PE250高通量测序,共得到有效序列257971条,质控过滤和去除嵌合体后得到219017条优质序列,聚类共得到2004个OTUs。各样品文库的覆盖度均在99.6%以上,并结合样品稀释曲线均趋于平缓,说明本研究测序数据合理,能够准确反映出土壤细菌群落的真实信息(图1)。3种退化草地土壤细菌群落丰富度指数(Chao1和ACE指数)依次为LDG>MDG>SDG(表4);细菌群落多样性指数(Simpson和Shannon-Wiener指数)依次为LDG>SDG>MDG。如图2所示,所有样品中共有OTUs数目为1317个,其中LDG、MDG和SDG中所特有的OTUs数目分别为112、100和47个。
2.4 不同退化草地土壤细菌群落分布特征分析
2.4.1门水平上的群落组成 通过测序发现,3种退化草地土壤细菌中相对丰度>1%的菌门为:放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)、酸杆菌门(Acidobacteria)、变形菌门(Proteobacteria)、芽单胞菌门(Gemmatimonadetes)、绿弯菌门(Chloroflexi)、疣微菌门(Verrucomicrobia)、浮霉菌门(Planctomycetes)、拟杆菌门(Bacteroidetes)和硝化螺旋菌门(Nitrospirae),丰度低于1%的类群占1.57%~3.07%。3种退化草地土壤细菌在门分类水平上,相对丰度存在一定的变化趋势(图3)。随着草地退化程度加剧,放线菌门、酸杆菌门、芽单胞菌门、绿弯菌门、浮霉菌门、拟杆菌门相对丰度呈先减小后增大趋势,厚壁菌门、疣微菌门和硝化螺旋菌门相对丰度呈先增大后减小趋势,变形菌门相对丰度呈逐渐减小趋势;功能菌群Proteobacteria/Acidobacteria呈先增大后减小的趋势。

表3 各样地土壤基本理化性质Table 3 The basic soil physiochemical indexes of different soil sampling sites
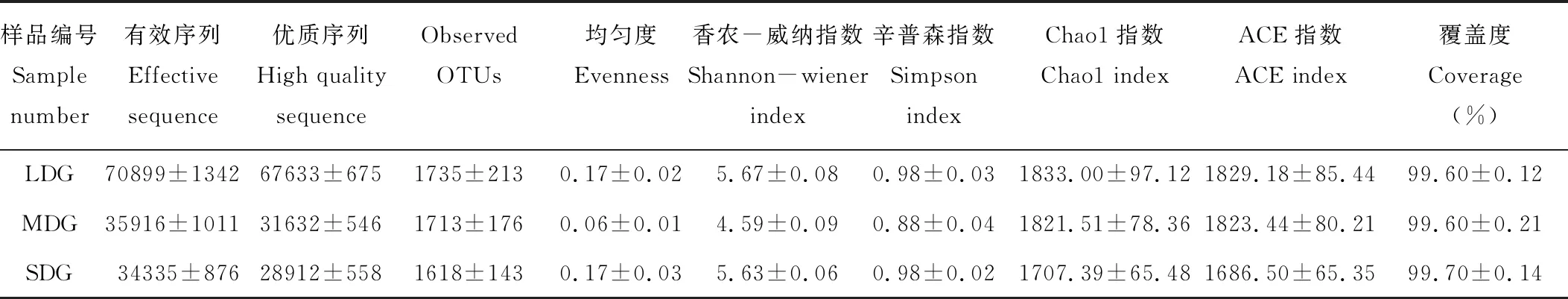
表4 样品序列数统计、丰富度与多样性指数Table 4 Sample sequence numbers statistics, richness and diversity index
2.4.2属水平上的群落组成 在属分类水平上,相对丰度>1%的菌属为:乳球菌属(Lactococcus)、土壤红杆菌属(Solirubrobacter)、未分类RB41属、Gaiella属、类诺卡氏属(Nocardioides)、假单胞菌属(Pseudomonas)、鞘氨醇单胞菌属(Sphingomonas)、未分类H16属、芽球菌属(Blastococcus)、Pseudarthrobacter、链球菌属(Streptococcus)和芽胞杆菌属(Bacillus),相对丰度低于1%的类群占48.94%~73.94%。3种退化草地土壤在属分类水平上,相对丰度存在一定变化趋势(图4)。随着退化程度加剧,乳球菌属、假单胞菌属、鞘氨醇单胞菌属、未分类H16属、Pseudarthrobacter和链球菌属相对丰度呈先增大后减小趋势,土壤红杆菌属、未分类RB41属、Gaiella、类诺卡氏属和芽球菌属相对丰度呈先减小后增大趋势,芽胞杆菌属相对丰度呈逐渐减小趋势。
2.5 Beta多样性分析
LDG与MDG样地间加权Unifrac距离相异系数分别为0.247和0.245(图5);LDG与SDG样地间加权Unifrac距离相异系数分别为0.184和0.216;MDG和SDG样地间加权Unifrac距离相异系数分别为0.294和0.258。结果表明,3种退化草地土壤间物种多样性差异为:LDG>SDG>MDG。为了研究不同样品间的相似性,对样品进行了UPGMA聚类分析。在门分类水平上,通过加权Unifrac距离的UPGMA聚类分析,LDG与SDG样地间细菌群落组成及丰度相似性较高,与MDG样地间细菌群落组成差异较大(图6)。
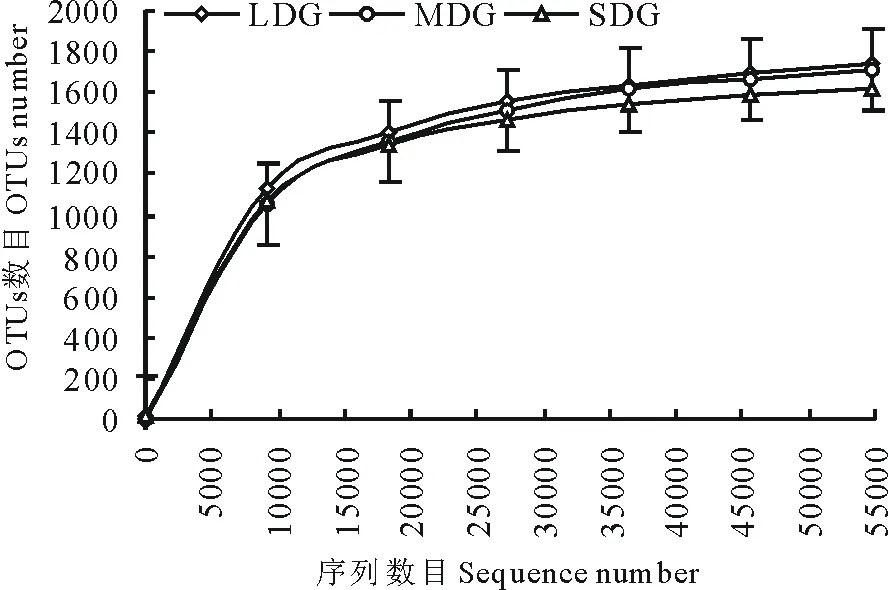
图1 样品稀释曲线Fig.1 Rarefaction curves for samples
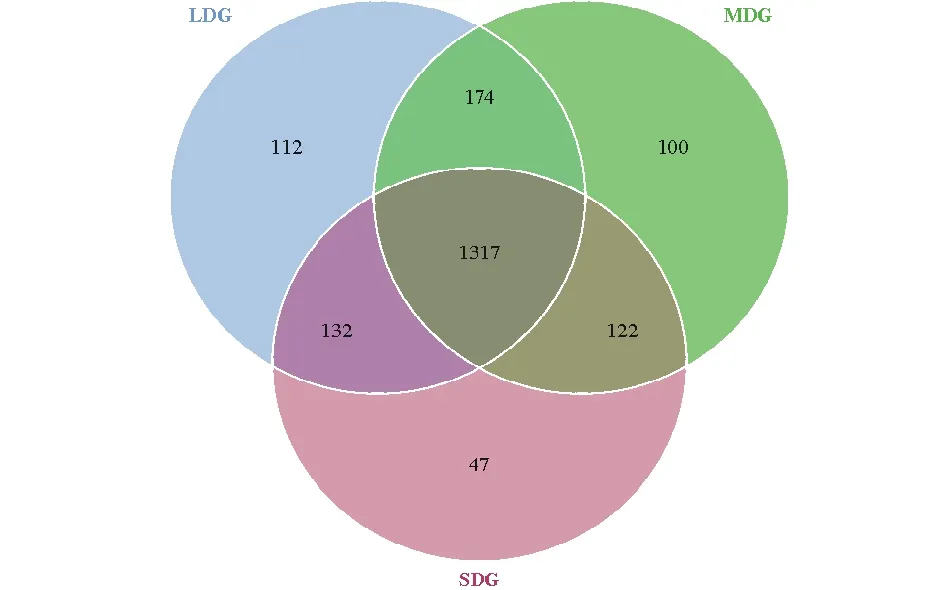
图2 样品韦恩图Fig.2 Venn diagrams of samples
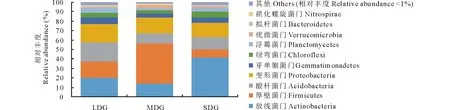
图3 门分类水平下的细菌群落相对丰度Fig.3 Relative abundance of bacterial community at phylum level
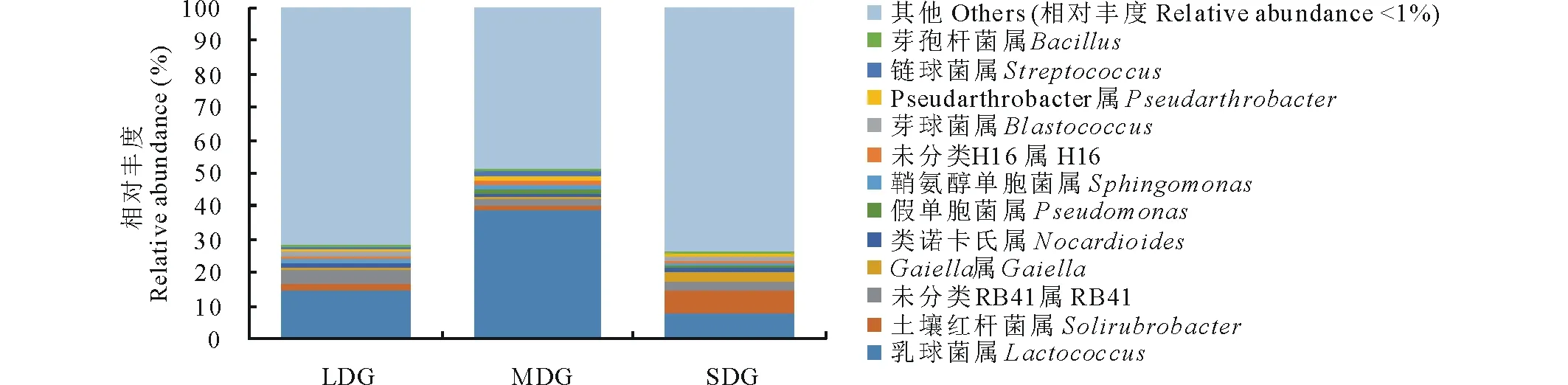
图4 属分类水平下的细菌群落相对丰度Fig.4 Relative abundances of bacterial community at genus level
2.6 细菌优势类群与土壤环境因子间RDA分析
通过RDA分析表明:第一、二排序轴累计解释率分别为68.1%和11.9%(图7)。其中,厚壁菌门与氧化还原酶呈显著正相关(P<0.05);放线菌门与中性磷酸酶、蔗糖酶、氧化还原酶、含水量呈极显著负相关(P<0.01),与电导率呈极显著正相关(P<0.01),与速效磷、速效钾呈显著负相关(P<0.05);变形菌门与脲酶、磷酸酶、脱氢酶、蔗糖酶、含水量、全N、全P、全K、速效N、速效P、速效K、有机质、有机碳呈极显著正相关(P<0.01),与pH、电导率和纤维素酶呈极显著负相关(P<0.01),与氧化还原酶呈显著正相关(P<0.05);酸杆菌门与脲酶、脱氢酶、全N、全P、全K、速效N、有机质、有机碳呈极显著正相关(P<0.01),与pH、纤维素酶呈极显著负相关(P<0.01),与蔗糖酶、速效P、速效K呈显著正相关(P<0.05);芽单胞菌门与pH呈极显著负相关(P<0.01);浮霉菌门与全P、速效N呈极显著正相关(P<0.01),与pH呈极显著负相关(P<0.01),与脲酶、全N、有机质、有机碳呈显著正相关(P<0.05);疣微菌门与pH呈显著正相关(P<0.05);硝化螺旋菌门与脲酶、全N、全P、速效N、有机质、有机碳呈极显著负相关(P<0.01),与pH呈极显著正相关(P<0.01);拟杆菌门与pH呈显著负相关(P<0.05)。综上,土壤酶活性和土壤理化性质均对细菌群落的分布均有影响,其中,土壤pH是土壤细菌群落分布的主要驱动因子。
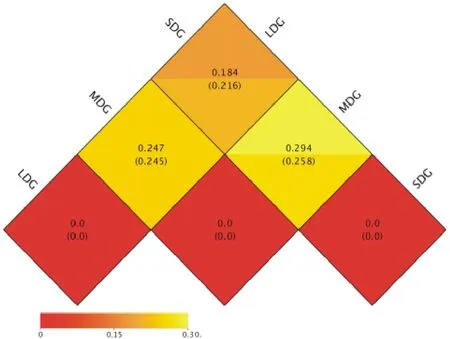
图5 各样地OTUs间的Heatmap分析Fig.5 Heatmap analysis between OTUs of sample plots level
3 讨论
草地植被退化和土壤理化性质的改变,将直接影响土壤微生物的生物量和群落结构多样性[24-27]。戴雅婷等[13]采用PCR-DGGE技术对内蒙古退化荒漠草原土壤细菌群落结构特征进行研究发现,随退化程度的加剧,细菌多样性指数由高到低依次为:轻度>重度>中度,本研究所得结果与其是一致的。本研究中相对丰度较高的细菌菌门为厚壁菌门、放线菌门、变形菌门和酸杆菌门,真菌菌门为子囊菌门和担子菌门,这与多数有关高寒草地土壤微生物群落组成所得研究结果是基本一致的。Li等[28]对青藏高原地区高寒草地土壤微生物群落结构组成进行研究发现变形菌门,放线菌门和酸杆菌门是该地区的优势种群。Zhou等[29]对青藏高原地区不同退化高寒草地土壤微生物群落结构组成进行研究发现放线菌门、变形菌门、酸杆菌门和绿弯菌门是该地区的优势种群。这表明不同高寒地区的退化草地土壤类型间微生物种群结构分布虽存在明显差异,但优势菌群基本相同。随着退化程度加剧,厚壁菌门整体上呈先升高后降低的趋势。这是因为厚壁菌门适于生长在可利用养分低的环境中,而高寒地区的低温环境又限制了有机物的可利用性,不利于它们的生长;另外,厚壁菌门在恶劣环境条件时进入休眠状态,尤其是能形成芽孢和孢子。随着退化程度加剧,放线菌门、酸杆菌门和变形菌门的丰度呈先减小后增大趋势,是由于高寒环境中土壤整体营养状况较低,且土壤pH属碱性导致的[30]。许多研究表明,在多种土壤生态系统中pH通常与细菌群落结构有显著的相关性,在近中性时多样性最高,pH一旦偏离中性,微生物群落会受到环境胁迫压力,多样性也因环境胁迫的选择而降低[31]。变形菌门是土壤中最主要的细菌类群,该类群的代谢活动是土壤中最主要的细菌活动。酸杆菌门一般存在于营养比较匮乏的环境,放线菌门能够降解纤维素和几丁质,是土壤养分供给的主要来源,并且产生的孢子能够抵抗外界不利的环境条件,使得它成为高寒草地土壤的优势菌群[32]。地上植被通过影响土壤中的有机碳、有机氮、土壤水分、温度、通气性和pH值等来影响土壤微生物多样性。植物群落多样性越丰富,凋落物和根系分泌物组成就越丰富,土壤微生物多样性也就越高[33]。植物多样性的增加往往伴随着植物生物量的增加,增加了凋落物和根系等有机物质的吸积,从而有利于土壤OC和N的积累[34]。本研究样地地处高寒环境,不同退化程度草地中土壤微生物群落结构的变化也相应地受到地上植被的影响,植被特征和细菌群落结构多样性显著相关(P<0.05),这说明地上植被群落结构的发展变化对细菌多样性产生影响。本研究发现,随着草地退化演替的发展,土壤细菌群落结构组成和多样性差异较大,这与植被和土壤理化性质是显著相关的。通过植被、土壤理化性质与细菌群落结构组成间的RDA分析发现,影响祁连山高寒草地土壤细菌群落结构组成的重要驱动因子是pH。大量研究也报道了pH对微生物的分布规律存在着普遍的影响[30]。本次研究也发现,pH对土壤微生物群落有显著性影响,但微生物种群对pH的响应是不同的,如土壤中酸杆菌门的丰度与pH值呈负相关,放线菌门的丰度与pH值呈正相关[30]。

图6 门水平上物种组成的UPGMA聚类树图Fig.6 UPGMA clustering tree diagram of species composition at the gate
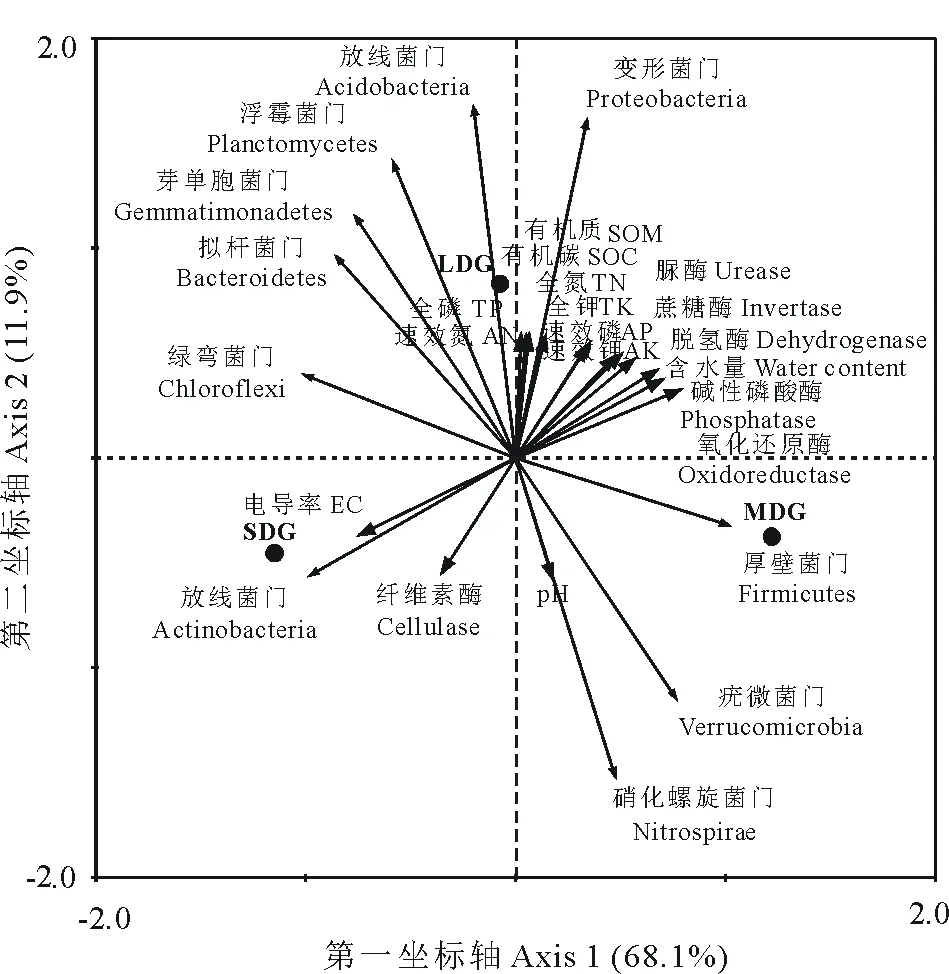
图7 细菌群落与土壤环境因子的冗余分析Fig.7 Redundancy analysis for bacterial community and soil environmental factors
4 结论
1) 植被盖度、高度、地上生物量和植物多样性指数随退化程度加剧均明显降低,依次为轻度退化>中度退化>重度退化;各样地土壤理化性质及酶活性间均差异显著(P<0.05)。
2) 通过高通量测序,共得到有效序列257971条,优质序列219017条,OTUs 2004个;土壤细菌群落丰富度指数表现为LDG>MDG>SDG;多样性指数表现为LDG>SDG>MDG;Beta多样性为LDG>SDG>MDG;3种退化草地土壤细菌群落的变化主要在于微生物量的变化,对细菌群落结构的影响并不明显。
3) RDA分析表明:土壤酶活性和土壤理化性质均对细菌群落的分布具有影响。其中土壤pH是影响土壤细菌群落分布的主要驱动因子。
猜你喜欢
现代园艺(2021年23期)2021-12-01 07:47:44
林业勘查设计(2020年1期)2021-01-18 02:40:48
新农业(2020年18期)2021-01-07 02:17:08
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
现代园艺(2017年21期)2018-01-03 06:41:42
绿色科技(2016年16期)2016-10-11 06:53:51
亚热带资源与环境学报(2015年1期)2015-01-22 07:04:58
