
三七总皂苷对羧酸酯酶体外活性的影响△
2019-07-13 03:47齐琪王彦莫雨佳罗菊元喻祥龙陆洋杜守颖
中国现代中药 2019年6期
齐琪,王彦,莫雨佳,罗菊元,喻祥龙,陆洋,杜守颖
北京中医药大学 中药学院,北京 102488
羧酸酯酶(Carboxylesterases,CESs),属于α/β水解酶超家族成员,是体内重要的Ⅰ相药物代谢酶,在不同种属动物体内普遍存在。人体内主要为羧酸酯酶1(hCE1)和羧酸酯酶2(hCE2),这两者氨基酸序列的同源性为48%,在组织分布、底物特异性等方面存在较大差异[1-2],hCE1在肝脏中高表达[3],hCE2在小肠和结肠中高表达[4]。人类与常用实验动物在hCE1和hCE2的分布上也存在显著的种属差异。羧酸酯酶除参与小分子脂肪酸和胆固醇的运输代谢外[5],还会直接影响药物的首过代谢(肠、肝),因药物导致相关羧酸酯酶的表达与活性变化是药-药相互作用的重要原因之一。如抗癌药伊立替康和卡培他滨等被设计成无活性的酯类前体药,经羧酸酯酶水解后起效,当联用白花前胡丁素(hCE1强抑制剂)[6]或五味子甲素(hCE2强抑制剂)[7]时,可使抗癌效果降低。又如抗血栓药氯吡格雷大部分在肝脏中经CESs代谢失活,剩下约15%经CYP酶系转化为活性代谢产物,CESs代谢能力的变化会影响氯吡格雷在体内的分布代谢,进而影响活性代谢产物的产生和药效的发挥[8]。
三七是一种散瘀止血常用中药,有很好的抗缺血损伤和抗脑缺血再灌注损伤活性[9],在心脑血管疾病中,常与化学药共用。如在阿司匹林与三七总皂苷相关制剂(血塞通、血栓通等)合用治疗脑卒中,两者联用后抗血小板凝集作用强于阿司匹林或三七总皂苷单用[10]。体内参与阿司匹林代谢的酯酶统称为阿司匹林酯酶,它主要作用于酯键的羧酸酯水解酶。阿司匹林原型药物可发挥药效作用,被水解为水杨酸后无抗血小板聚集作用。本课题组前期研究发现,三七总皂苷可降低阿司匹林的体内代谢,这可能是联用增效的原因之一。为进一步明确三七总皂苷对阿司匹林的作用机制,本实验在体外肝微粒体水平下对三七总皂苷抑制羧酸酯酶活性的能力进行探究,为三七联用羧酸酯酶底物提供合理的科学依据。
1 材料与仪器
1.1 材料
荧光素二乙酸酯(FDA)和荧光素(F)(上海阿拉丁生化科技股份有限公司,纯度>97%),2-(2-苯甲酰基-3-甲氧基苯基)-苯并噻唑(BMBT)和其水解产物(HMBT)(大连化学物理研究所,纯度>98%),双(4-硝基苯基)磷酸酯(BNPP)(东京化成工业株式会社,纯度>98%),三七总皂苷原粉(PNS)(云南三七科技有限公司,批号:110870-201603),小鼠肝微粒体(MLM)(美国Corning Gentest公司),二甲基亚砜(DMSO)(北京化工厂,分析纯),乙腈[赛默飞世尔科技(中国)有限公司,色谱纯],磷酸盐缓冲液(PBS)(美国Corning公司,1×,pH 7.4)。
1.2 仪器
BS224型电子天平(德国赛多利斯公司),微量移液器(德国艾本德公司),307057型空气浴振荡摇床(江苏太仓豪诚实验仪器制造有限公司),5427型高速离心机(德国艾本德公司),SpectraMax i3x型多功能酶标仪(美国Molecuar Devices LLC公司)。
2 方法与结果
2.1 方法
在200 μL的反应体系中,含有PBS缓冲液(100 mmol·L-1,pH 7.4)及适量的小鼠肝微粒体,涡旋混合均匀后,于37 ℃恒温空气浴共同振荡孵育活化10 min,向体系中分别添加适量的荧光探针BMBT(hCE1特异性底物)和FDA(hCE2特异性底物)起始反应,37 ℃振荡孵育20 min后,加入等体积冰乙腈涡旋终止反应,8000 r·min-1离心3 min去除蛋白。移取200 μL上清液至96孔板,于荧光酶标仪测量荧光强度(FDA水解产物F激发波长为480 nm,发射波长为520 nm;BMBT水解产物HBMT激发波长为304 nm,发射波长为488 nm)。
2.1.1 反应条件优化
2.1.1.1 底物浓度优化 肝微粒体浓度保持2 μg·mL-1(BMBT),0.2 μg·mL-1(FDA),将底物浓度设为1、2、5、10、20、30 μmol·L-1;酶活化后加入探针底物,反应2 min,每15 s测定荧光强度,时间和荧光强度线性拟合得到的斜率即为反应速率,将底物浓度和反应速率代入米氏方程进行非线性拟合得到动力学常数Km。
2.1.1.2 肝微粒体浓度优化 采用接近Km的底物浓度,将肝微粒体质量浓度设为0.25、0.5、1、2、3 μg·mL-1(底物为BMBT),0.1、0.15、0.2、0.3、0.5 μg·mL-1(底物为FDA);按2.1方法测量荧光强度,绘制酶浓度-荧光强度曲线。
2.1.1.3 DMSO用量优化 采用优化好的底物浓度和肝微粒体浓度,将溶解探针底物的DMSO用量设为0%、1%、2%、3%(V/V);按2.1方法测量荧光强度,按公式(1)计算羧酸酯酶残余活性。

(1)
2.1.1.4 乙腈用量优化 采用优化好的底物浓度和肝微粒体浓度,在活化时分别加入0%、50%、75%、100%(V/V)的冰乙腈,再按2.1方法离心后向反应体系中补加冰乙腈,使冰乙腈终体积均为200 μL,取200 μL上清液至96孔板,于荧光酶标仪测量荧光强度,计算羧酸酯酶残余活性。
2.1.2 三七总皂苷对羧酸酯酶的抑制作用 采用优化好的酶促反应条件,将三七总皂苷质量浓度设为1、10、100 μg·mL-1,与酶共孵育活化,按2.1方法测量荧光强度,并公式(2)计算对羧酸酯酶活性的抑制率。阴性对照(等体积PBS代替三七总皂苷)和阳性对照(等体积羧酸酯酶阳性抑制剂BNPP,终浓度100 μmol·L-1代替三七总皂苷)在相同条件下反应。选择抑制效果较为明显的羧酸酯酶亚型,将三七总皂苷质量浓度设为1、5、10、20、50、100、300、500、700 μg·mL-1,计算三七总皂苷对羧酸酯酶活性的抑制率,并计算IC50值。
抑制率=1-残余活性
(2)
2.1.3 抑制类型的判断 选择5组三七总皂苷质量浓度,采用优化好的酶促反应条件,将三七总皂苷与肝微粒体共孵育活化,按“底物浓度优化”方法测量动力学常数(Km)和最大反应速率(Vmax),转化为双倒数方程,判断抑制类型。
2.1.4 体外-体内外推 通过抑制剂浓度和相对应的双倒数方程的斜率计算求得抑制动力学参数Ki;体外-体内外推使用的方程为:AUCi/AUC=1+C抑制剂invivo/Ki,若C抑制剂invivo/Ki<0.1,无药物相互作用可能性;0.1
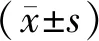
2.2 结果
2.2.1 最适反应条件
2.2.1.1 最适底物浓度 通过米氏方程非线性拟合(如图1),当底物为BMBT时,获得的动力学常数Km=(12.79±2.427)μmol·L-1,BMBT最适浓度为13 μmol·L-1;当底物为FDA时,获得的动力学常数Km=(7.866±1.367)μmol·L-1,FDA最适浓度为8 μmol·L-1。
注:A.BMBT;B.FDA。图1 小鼠肝微粒体中BMBT(A)/FDA(B)水解产物的动力学行为
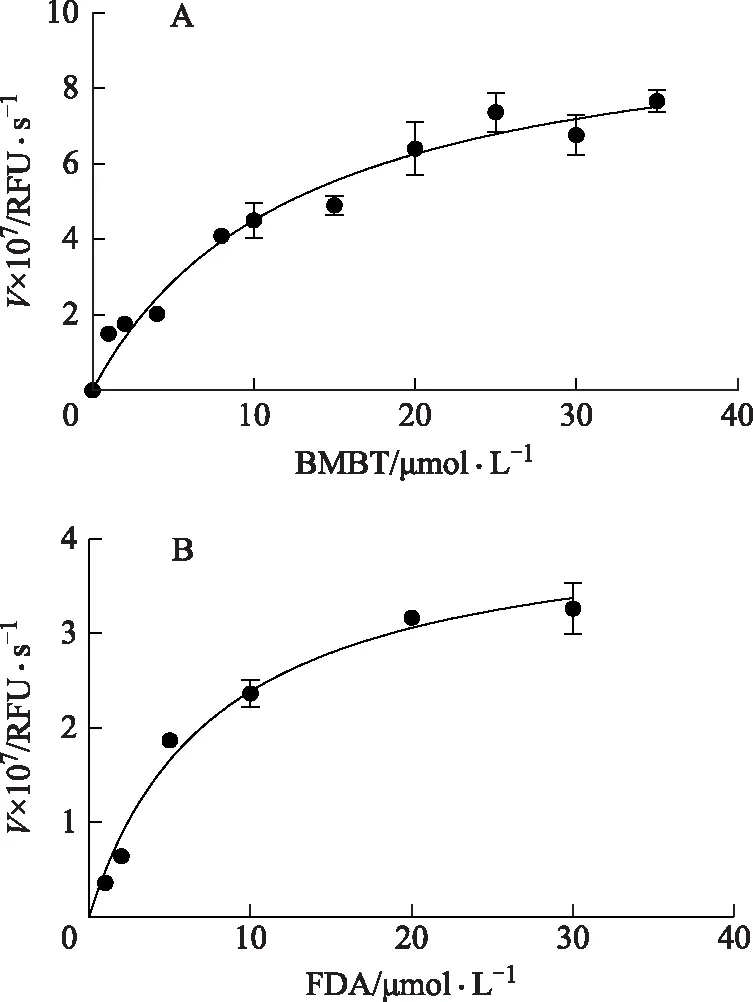
2.2.1.2 最适肝微粒体浓度 为保证实验结果准确,底物水解率不得高于20%。当底物为BMBT时,荧光强度(Y)与肝微粒体浓度(X)回归方程表示为Y=380 120X-34 674(r=0.996 0),BMBT的最适肝微粒体浓度为2 μg·mL-1;当底物为FDA时,回归方程表示为Y=6.40×108X-1.10×107(r=0.996 5),FDA的最适肝微粒体浓度为0.2 μg·mL-1。见图2。
注:A.BMBT;B.FDA。图2 BMBT(A)/FDA(B)水解产物荧光强度随肝微粒体浓度的线性变化
2.2.1.3 有机溶剂用量 结果表明,当体系中存在0%~3% DMSO时,hCE1和hCE2的活性没有显著变化;体系中加入一半体积以上的冰乙腈可以终止酶促反应,最终确定加入200 μL冰乙腈。
2.2.2 三七总皂苷对羧酸酯酶的抑制作用 如图3所示,当质量浓度为100 μg·mL-1时,三七总皂苷对hCE1的抑制率仅为27%;而三七总皂苷质量浓度为10 μg·mL-1时,对hCE2的抑制率达到42.1%,当质量浓度为100 μg·mL-1时,抑制率为63.5%,抑制效果较为明显(见图4),进一步细化三七总皂苷浓度进行抑制率实验,求得三七总皂苷对hCE2的半数抑制率IC50为17.05 μg·mL-1。

注:A.hCE1;B.hCE2;NC代表阴性对照;PC代表阳性对照。图3 三七总皂苷对hCE1(A)/hCE2(B)活性抑制效果的初筛
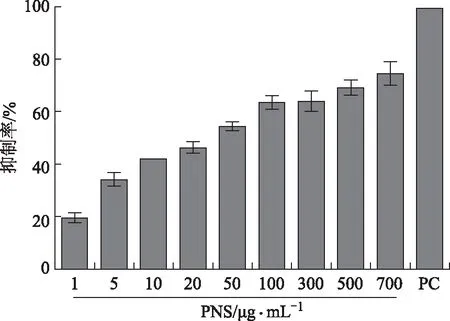
图4 三七总皂苷对hCE2活性的抑制作用
2.2.3 抑制类型 通过双倒数方程对实验结果进行分析,所得的双倒数拟合直线相交于X轴(如图5),说明三七总皂苷对hCE2的抑制类型为非竞争型抑制;将各斜率和对应的三七总皂苷浓度进行方程拟合,所得函数为Y=0.003 7X+0.29,进一步计算求得三七总皂苷对hCE2的Ki为77.53 μg·mL-1。
2.2.4 体内药-药相互作用的发生可能性 大鼠在灌胃三七总皂苷(600 mg·kg-1)后,采用整合药代动力学的方法,拟合出三七总皂苷的Cmax为18.473 μg·mL-1[12],将此浓度(C抑制剂)代入体外-体内外推方程,求得C抑制剂/Ki值为0.24,这表明由于抑制hCE2活性所造成的药物生物利用度比未服用三七总皂苷时增大了0.24倍。
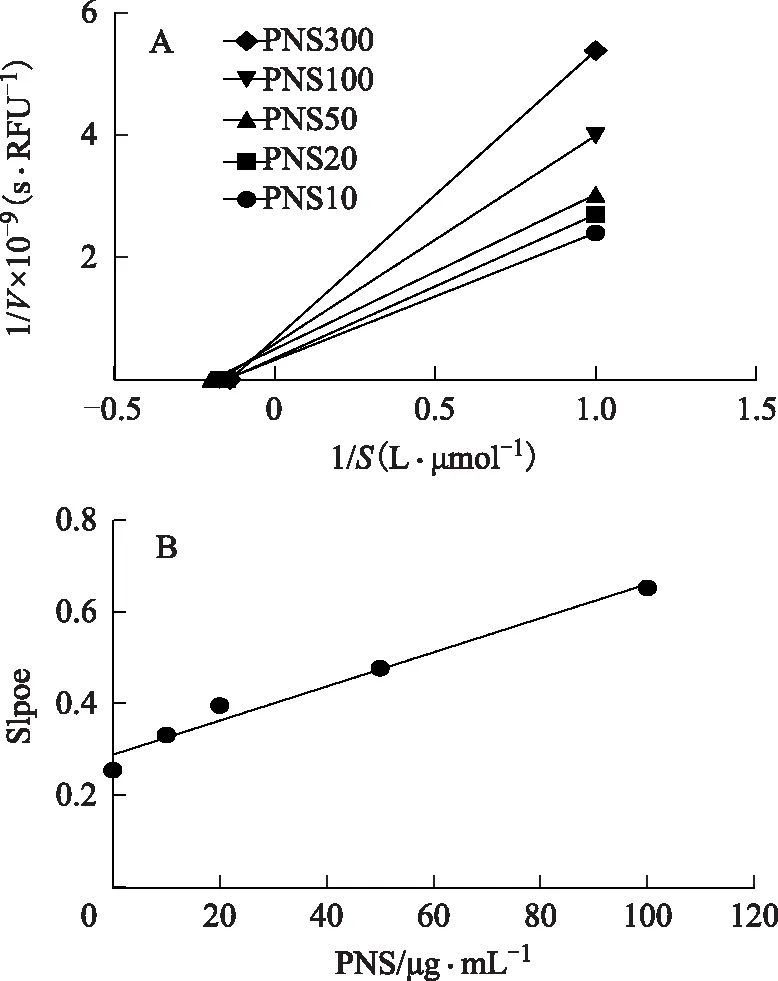
注:A.双倒数方程;B.抑制动力学参数方程。图5 三七总皂苷对hCE2活性抑制的双倒数方程和抑制动力学参数方程
3 讨论
本研究证明,三七总皂苷可较明显地直接抑制hCE2的水解活性,且通过体外-体内外推方法证明,三七总皂苷在与经hCE2代谢的药物一起服用时,需要警惕药-药相互作用的发生。在体外水平下,三七总皂苷对hCE1的活性表现出微弱的直接抑制作用,但是三七总皂苷对hCE1的活性是否有间接抑制作用还需要进一步考察:由于选择肝微粒体作为酶源,并不存在羧酸酯酶的表达翻译过程,但三七总皂苷还有可能通过影响hCE1的转录翻译环节,从而抑制其水解活性。
羧酸酯酶的催化中心由一个包含丝氨酸催化三联体的中央催化位点组成,丝氨酸残基很容易与含羰基分子反应形成中间体,若中间体的碳碳双键无法断裂,则会抑制催化活性。具有两个相邻羰基结构1,2-二酮类分子大多可强烈抑制羧酸酯酶的活性[13],而三七总皂苷中所含有的5种皂苷(三七皂苷R1、人参皂苷Rg1、人参皂苷Re、人参皂苷Rb1、人参皂苷Rd)均不具有这种可以和活性催化中心结合的相邻羰基结构,说明三七总皂苷对羧酸酯酶的抑制类型不可能是竞争型抑制,本实验进一步研究发现三七总皂苷对hCE2的抑制类型为非竞争型抑制,说明三七总皂苷与hCE2的非活性部位相结合,形成抑制物-酶的络合物后进一步再与底物结合;或是hCE2与底物结合成底物-酶络合物后,其中有部分再与三七总皂苷结合。这两种情况所形成的中间络合物都不能直接生成水解产物,从而导致了酶催化反应速率的降低。
三七作为一种中长期服用的中药,在肠道中对羧酸酯酶的抑制效果可能是累积作用。因此,长期服用三七总皂苷,同时使用羧酸酯酶底物药物时应该建议进行体内药物水平的监测,必要时可调整用药量,以确保临床安全有效。
猜你喜欢
轻工学报(2022年3期)2022-06-22
黑龙江八一农垦大学学报(2021年6期)2021-12-30
昆明医科大学学报(2021年6期)2021-07-31
建材发展导向(2021年7期)2021-07-16
建材发展导向(2021年24期)2021-02-12
农产品加工(2020年23期)2021-01-11
建材发展导向(2019年5期)2019-09-09
分析化学(2017年12期)2017-12-25
中国民族民间医药·上半月(2017年2期)2017-03-09
科学中国人(2015年2期)2015-07-26
