
基于电化学寡核苷酸传感器的痕量汞离子检测*
2019-07-08 09:10孙嘉弟王心怡
传感技术学报 2019年6期
孙嘉弟,甘 颖,梁 韬,王心怡,万 浩,王 平
(浙江大学生物传感器国家专业实验室,生物医学工程教育部重点实验室,生仪学院,杭州 310027)
水是人类生存必不可缺的资源,但近年来国内外各类水体的水质却面临各方面的考验,重金属汞污染就是主要威胁之一。汞以元素汞、无机汞和有机汞三种形式存在于自然水体环境中[1]。工业汞污染排放以无机汞为主[2],就算仅有少量汞离子进入到血液中,它也极容易与肾细胞的蛋白质牢固结合并具有抗原性,引起变态反应,使得肾功能遭受严重破坏而引起肾病综合症[3]。汞离子还能在微生物的转化作用下转变成有机汞,有机汞难降解,会在水生生物体内积蓄,通过食物链富集进入人体后将对健康造成严重损害。人体内多数功能性酶均为蛋白质,蛋白质由氨基酸通过巯基相连组成,而汞能与巯基形成稳定的络合物而使酶蛋白失活,且还能与羧基、氨基和磷酰基这些功能性基团络合而破坏其活性,进而阻碍细胞的生物活性及正常代谢,导致各种病变[4]。然而,汞及其化合物由于有很多特殊性能,已被大规模地应用到生产中,我国目前涉及汞的行业众多,由此造成的污染范围非常广,且有逐步扩大的态势,我国汞污染问题会在相当长一段时间内存在着[5]。因此,在环境和食品监测领域,具有高灵敏度的汞离子检测方法受到广泛关注。
传统的汞离子检测手段有原子吸收光谱法(AAS)、原子发射光谱法(AES)及等离子体电感耦合质谱法(ICP-MS)等[6]。AAS的原子吸收带宽很窄,虽然选择性较强,但对样品制作的要求较高,对复杂样品的分析精密度较低,分析时间较长,对设备的要求也高,而且无法同时分析多种不同的元素[7];AES分析速度快、选择性好,可同时分析多种元素,与其他方法的结合也受到广泛关注,但所用设备昂贵,当元素的含量较高时,其分析误差较大,而当元素为超微量级时,分析的灵敏度又不够;ICP-MS[8]检出限低,被公认为最强有力的痕量元素分析技术,其检测范围宽,灵敏度高,还可进行多元素分析,但是由于检测过程中需要将样品汽化,这就要求样品热敏性低,溶液中悬浮杂质要少,即对待测样品的要求较高,更主要的是仪器造价昂贵,且不能进行即时检测。这些传统检测手段所需仪器昂贵而笨重,而且样品预处理过程繁琐,并不适用于现场痕量检测[9],而另一些新兴的方法,如冠醚类汞离子传感器[10]、表面增强拉曼光谱法及阳极溶出伏安法[11],虽然相对而言更经济便捷,但也存在选择性不高的缺点。
相比以上方法,电化学传感器检出限低,灵敏度高,操作简便,且易于微型化,方便设计成便携式仪器,是最适合用于现场快速检测的传感系统,基于寡核苷酸构建的电化学传感器还具有高度特异性,具有非常广的应用前景。因此本文构建了一种基于T-Hg2+-T特异性结构的电化学寡核苷酸传感器,利用循环伏安法(CV)及交流阻抗谱法(EIS)等电化学分析手段在实验室环境下对溶液中的Hg2+浓度进行了定量检测与分析,通过优化固定方式、固定时间以及固定比例提高了其性能指标,成功检测到痕量汞离子,为现场监测痕量汞离子提供了一种新的解决方案。
1 材料与方法
1.1 试剂和仪器
寡核苷酸链5′-S-S-(CH2)6-TTTTT TTTTT TTTTT TTTTT-3′(T20-SH)购自南京金斯瑞生物科技有限公司。6-巯基-1-己醇(MCH)和三(2-羧乙基)膦盐酸盐(TCEP)购自阿拉丁工业公司,TE缓冲液购自美国Sigma-aladin公司。汞离子标准液购自国家标准物质资源平台。配溶液用水均为由Milli-Q纯水系统制得的超纯水(≥18.3 MΩ)。实验所用仪器CHI660E电化学工作站购自上海辰华仪器有限公司,KQ3200DE数控超声波清洗器购自昆山超声仪器有限公司,ThermoFisher离心机购自上海百基生物科技有限公司。
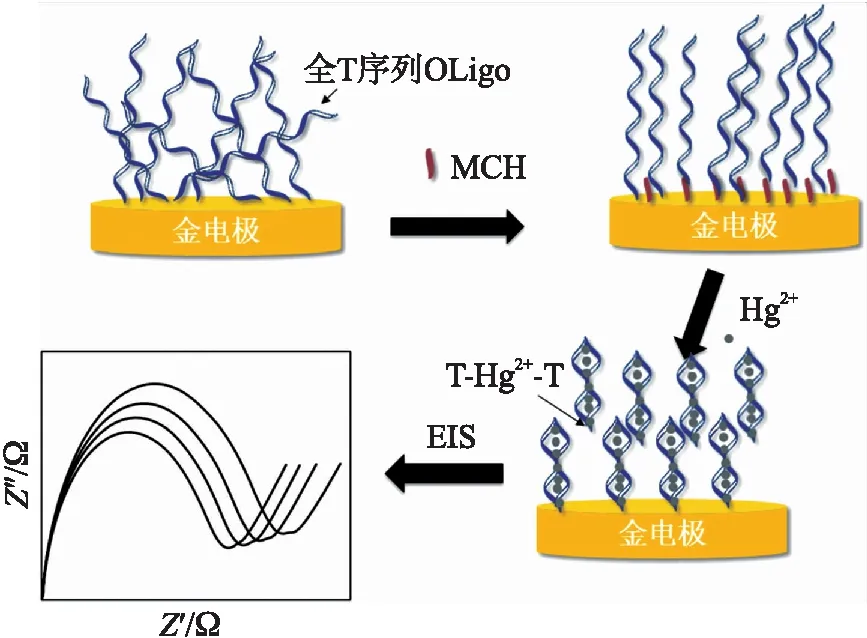
图1 电化学寡核苷酸传感器检测Hg2+示意图
1.2 检测原理及仿真模型
1.2.1 检测原理
检测原理如图1所示。全T序列寡核苷酸链(Oligo)的5′端修饰巯基,可以通过Au-S键的共价键合作用自组装到金电极表面。Oligo结构柔韧,容易弯曲,而且除了Au-S键外还可以通过Au-N键与金电极结合,碱基暴露在外侧很容易吸附到金电极的表面,这种非特异性吸附使得单链不规则地平躺在金电极表面。经过MCH封闭后,MCH通过竞争Au-S键结合位点来消除非特异性吸附,这种小分子自组装层保证Oligo能够保持一致而稳定的结构固定在金电极表面。当检测到Hg2+时,Oligo中的胸腺嘧啶与Hg2+特异性结合后形成一种“发卡型”结构,这种构象上的改变增强了相邻链间的空间位阻和库仑力,导致电极表面部分Oligo的释放[12]。金电极表面的Oligo形态和密度上的变化能够引起双电层电容和电子转移阻抗的变化,而后者的变化量可以通过交流阻抗谱法来定量测定。样品Hg2+浓度不同,Oligo形态和密度变化的程度不同,对应的电化学指标改变量也不同,由此来定量检测溶液中的汞离子浓度。
1.2.2 仿真模型
对实际得到的Nyquist图构建等效电路进行仿真。首先构建该电池的等效电路,如下图2电路模型所示。已知圆弧是由RC元件并联组成的,且圆弧直径大小等于生成它的RC元件中电阻R的阻值。放大上述Nyquist图的高频部分,在实际图线中大圆弧并非与横轴直接相交得到表征溶液电阻RS的截距,而是在大圆弧之前还存在一个非常小的半圆弧,表明溶液除阻抗成分外还存在电容成份,因此在RS并联C1。实际中扩散层每层的离子浓度都不同,不过每层的阻抗行为等效于RC元件的并联,为简化电路,这个RC元件的无穷集合就用Warburg阻抗W1表示,双电层的阻抗行为就用RCT、W1和双电层电容的并联来等效。
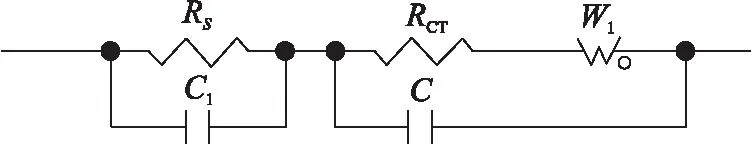
图2 仿真模型
1.3 方法与步骤
清洗工作电极:金电极作为自组装膜的基底,其表面的清洁程度是Oligo自组装膜形成与否、稳定与否的关键。预处理中先将工作电极在Piranha溶液中浸泡30s,去除电极表面的有机物。然后在金属电极专用抛光海绵上依次用1.0 μm,0.3 μm,0.05 μm的氧化铝粉末研磨抛光,按“8”字形沿顺、逆时针各研磨50圈。再进行超声清洗,依次在无水乙醇和去离子水中超声5 min。最后进行电化学清洗,将三电极系统浸没到0.5 mol/L稀硫酸中,进行循环伏安(CV)扫描直至CV曲线达到稳定且出现较好的金氧化还原峰。取出电极,先用超纯水清洗再用氮气吹干。
配制探针溶液:Oligo冻干粉先经12 000 r/min高速离心1 min再用TE缓冲液配成100 μmol/L的Oligo溶液,密闭后充分震荡混匀。加TCEP断开二硫键:分别取2 μL上述Oligo溶液及100 μmol/L MCH溶液至离心管,各自加入等量100 mmol/L的TCEP,充分震荡后静置30 min,防止Oligo及MCH中巯基氧化形成二硫键混合物。配制Oligo-MCH-TCEP混合液:取2 μL配好的MCH-TCEP加到Oligo-TCEP混合液中,用0.01 mol/L PBS稀释,充分震荡并混匀。
修饰及表征:将金电极表面浸没到上述Oligo-MCH-TCEP混合液中,在4 ℃的冰箱中静置8 h。固定完毕后取出金电极,先浸在TE缓冲液中静置20 min,除去未固定上的Oligo。然后将三电极体系浸没到16 mL 2 mmol/L K3[Fe(CN)6]/K4[Fe(CN)6](含0.1 mol/L KCl)混合液中,扫描并记录金电极经过表面修饰后的交流阻抗曲线。
检测样品:让金电极表面完全浸没在汞标准液中,在室温下反应10 min,用PBS缓冲液清洗后,把三电极体系一起浸没到16 mL 2 mmol/L K3[Fe(CN)6]/K4[Fe(CN)6](含0.1 mol/L KCl)中,扫描并记录交流阻抗曲线。单次检测结束后取出金电极,用PBS缓冲液清洗后继续与汞标准液反应,并逐次加大汞标准液浓度,检测电化学阻抗的变化。另设一空白对照组。
1.4 优化实验
金电极表面Oligo修饰层的有序性、完整性和稳定性不仅受到表面清洁度的影响,还与Oligo的固定方式、固定时间、与MCH的浓度比例等因素相关,为尽可能地提高传感系统的性能指标,在保证金电极表面清洁度较理想的前提下,本研究进行了系列优化实验。
固定方式的优化:根据Oligo固定跟MCH封闭这两个步骤的前后顺序,可把Oligo的固定方式分为预固定、共固定和后固定三种。预固定就是在金电极表面先自组装上Oligo,然后加MCH进行封闭,用较强的Au-S键取代较弱的Au-N,消除非特异性结合;共固定是将金电极浸没在Oligo和MCH的混合溶液中,让两者竞争Au-S结合位点,同时固定到金电极表面;后固定则是先进行MCH封闭,在金电极表面形成小分子自组装层,再固定上Oligo,让Oligo的Au-S键去取代MCH的Au-S键。通过阅读文献,研究均表明后固定的效果不理想,故本研究着重比较了预固定和共固定两者的效果。
固定时间的优化:设置了固定时间分别为4 h、8 h、12 h和16 h的四组对照实验,均采用共固定方式以及1∶1的Oligo和MCH比例,同组PBS缓冲液先后测试8次,用交流阻抗法对金电极表面Oligo的固定量及稳定性进行检测分析。
固定比例的优化:实验中设置了三组对照,混合液中Oligo与MCH的浓度比依次为1∶1、1∶10、1∶100,并用交流阻抗法进行了检测分析。
1.5 特异性实验
为验证构建的传感器对Hg2+的选择性,本研究还进行了特异性实验,即在相同条件下用这一传感器检测其他重金属离子,对比检测效果。实验中设置了七组对照,分别为等体积(100μL)的10 nmol/L Hg2+标准液、1 μmol/L Cu2+标准液、1 μmol/L Cd2+标准液、1 μmol/L Pb2+标准液、1 μmol/L Cr3+标准液、超纯水、10 nmol/L Hg2+与1 μmol/L所有其余离子的混合液。
2 结果与讨论
2.1 工作电极表征
如图3所示,金电极上修饰了Oligo后,在CV图中其电流明显减小,在Nyquist图中其电子转移阻抗大大增加,这是因为Oligo双螺旋骨架上的磷酸基团水解后带负电,电极表面增加的负电荷阻碍了电子传递,也阻碍了[Fe(CN)6]3-和[Fe(CN)6]4-的扩散。而经过MCH封闭处理之后,金电极的电子转移阻抗略有减小,这是因为带有巯基的MCH能以Au-S键与金电极结合,且Au-S键的键能大于Au-N键,MCH可以取代与金电极非特异性结合的Oligo。此外,MCH小分子组装层还可以使得Oligo竖立在金电极表面,修饰层更加有序规整,电子传递和阴离子的扩散相对容易一些。

图3 金电极修饰前后的表征图
2.2 优化实验
两种固定方式下的阻抗值及其变化率如图4(a),结果表明在相同条件下,采用共固定方式固定上的Oligo明显多于预固定方式,且检测完同浓度的汞标准液后,共固定和预固定的阻抗变化率分别为79%和88%,前者的变化更明显。重复实验得到了一致的结果。共固定方式效果更佳可能是因为固定过程中Oligo与MCH同时竞争金电极表面的结合位点能使自组装膜更均匀。

图4 优化结果
如图4(b),结合固定量和稳定性来分析,固定4 h,虽然金电极表面的电子转移阻抗在固定前后的变化量最大,看似固定量最高但是非常不稳定,这可能是因为4 h内MCH小分子自组装层还没有形成稳定,多数Oligo仍不规则地平躺在金电极表面,导致固定量“虚高”,且非特性结合并不稳定,检测时交流阻抗的波动较大。固定8 h后,金电极表面电子转移阻抗的变化量较小,即修饰层较为稳定,此时MCH小分子自组装层达到稳定状态。固定时间为12 h,金电极表面电子转移阻抗的变化量又有明显下降,修饰层稳定性也变差,这可能是随着时间的加长,有部分Oligo自然地脱落下来。当固定时间再加长到16 h时,金电极表面电子转移阻抗的变化量略有下降,而且修饰层达到了非常稳定的状态,此时电极表面留下结合最为牢固的Oligo,很少再有脱落。固定时间为16 h,虽然修饰层的稳定性最好,但是电极表面固定上的Oligo太少,能检测的Hg2+浓度范围非常有限。而固定时间为8 h,其固定量与稳定性均较为可观,因此综合比较之下,选定最佳固定时间为8 h。
如图4(c),将阻抗值归一化后,建立阻抗变化率与汞离子浓度间的线性关系,发现阻抗变化率与汞离子浓度的对数值间呈线性。比较各固定比例之间的结果可以发现,当Oligo∶MCH=1∶1时,斜率最大,响应最灵敏。而当Oligo∶MCH=1∶100时,线性范围、线性度及响应灵敏度均不理想,这可能是因为MCH浓度过高,过多地竞争到Au-S结合位点,导致金电极表面能固定上的Oligo太少。
2.3 特异性实验
检测前后阻抗变化率的对比结果如图5所示,传感器对10 nmol/L Hg2+标准液和对所有离子混合液的响应近乎一致,即对混合液的响应基本来自Hg2+,而对1 μmol/L其余金属离子的响应与超纯水的近乎一致,这一结果表明本研究构建的传感器对Hg2+具有高度选择性。
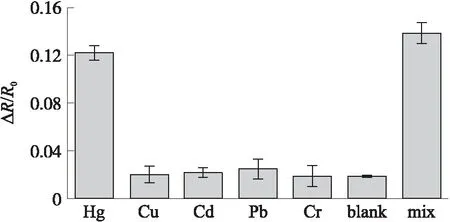
图5 传感器对10 nmol/L Hg2+标准液、1 μmol/L 干扰离子、超纯水及混合液的响应结果
2.4 标准曲线
根据以上优化实验结果,以1∶1的Oligo和MCH浓度比在4 ℃环境下共固定8 h后构建出传感器,然后用于检测一系列浓度的汞标准液,绘制对应的Nyquist图,并进行三次重复实验。进行Randles电路仿真,计算每条图线对应的电子转移阻抗RCT,分析RCT与汞离子浓度之间的定量关系。仿真结果示意图如图6所示,可见利用采用的等效电路模型得到仿真结果与原图线有较高的吻合度。
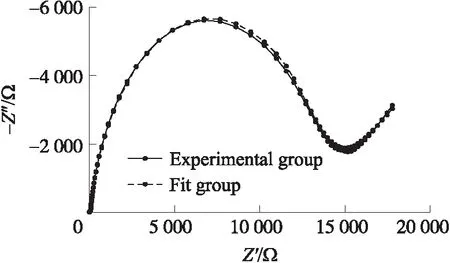
图6 仿真结果
与不同浓度汞液反应后金电极的Nyquist图如图7(a)所示,曲线从a到g对应的汞液浓度依次为0.5 nmol/L,3 nmol/L,10 nmol/L,50 nmol/L,100 nmol/L,500 nmol/L,2 μmol/L。结果表明当用所构建的这一传感器去检测一系列浓度的汞液时,RCT随Hg2+浓度的增加依次减小。这是因为金电极表面Oligo与Hg2+特异性结合后形成一种“发卡型”结构,导致相邻Oligo序列间的空间位阻和库仑力增强,电极表面部分Oligo分子释放,密度减小,电子传递更容易,RCT也因此减小,Hg2+浓度越高结合得越多,RCT减小越多。
对阻抗值进行归一化处理,最小二乘法线性拟合后得到如图7(b)所示阻抗变化率与浓度对数值之间的线性关系。本传感器的线性范围为0.5 nmol/L~500 nmol/L,检出限为0.2 nmol/L,且具有较好的重复性。
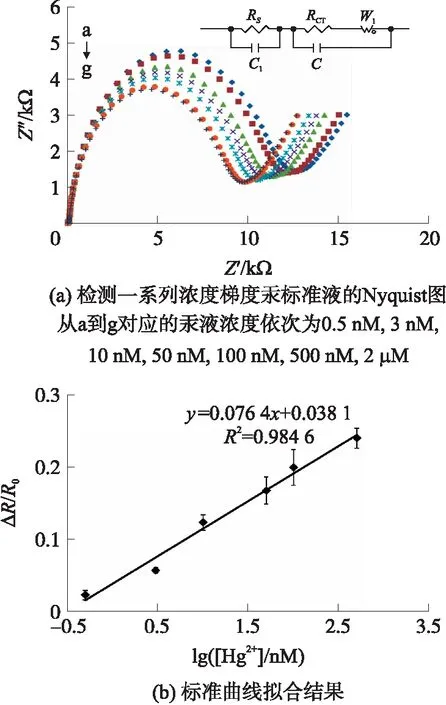
(a)检测一系列浓度梯度汞标准液的Nyquist图,从a到g对应的汞液浓度依次为0.5 nmol/L,3 nmol/L,10 nmol/L,50 nmol/L,100 nmol/L,500 nmol/L,2 μmol/L;(b)标准曲线拟合结果图7 检测结果图
2.5 回收率检测
为了评估所构建的这一传感器的适用性和可靠性,我们对饮用水进行了检测,饮用水本底背景检测结果如表1所示,由于饮用水中汞含量极低,利用ICP-MS也无法测得,于是我们检测了实际水样加入不同浓度Hg2+后的回收率,结果如下表2所示,加标浓度为0.5 nmol/L、1 nmol/L和5 nmol/L,其回收率分别为92%、125%和97%。以上结果表明,我们所构建的这种传感器用于检测饮用水中的微量汞离子时有较为令人满意的结果。
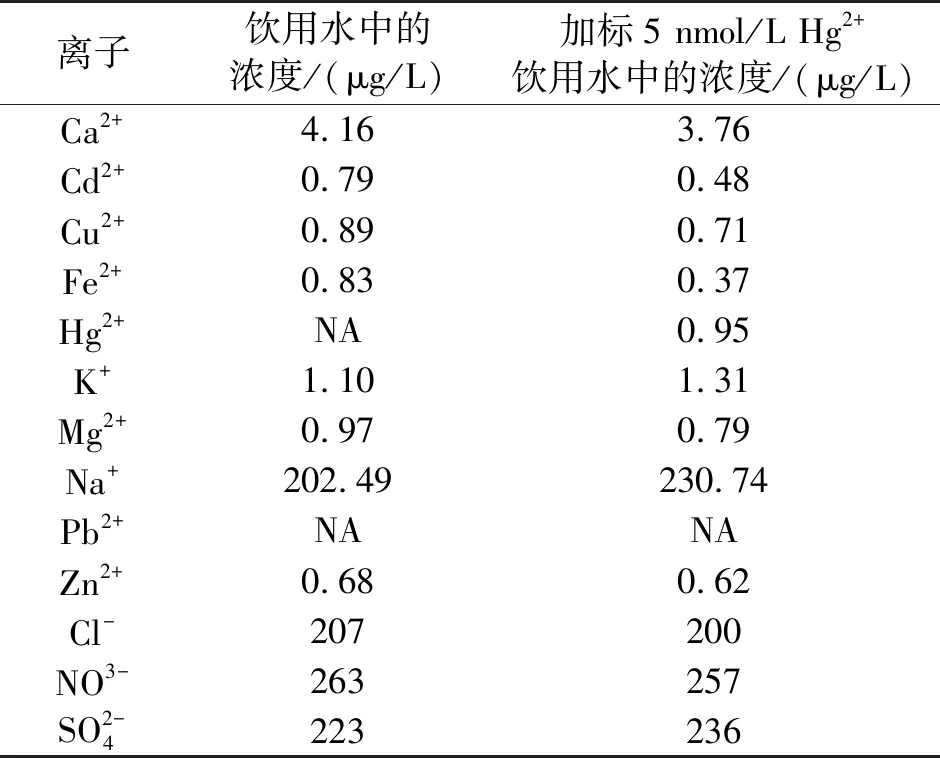
表1 饮用水加标前后的离子浓度ICP-MS测定结果

表2 饮用水加标后的Hg2+回收率
3 结论
本文构建了一种检测痕量Hg2+的电化学寡核苷酸传感器,通过对固定方式、固定时间以及固定比例三方面进行优化,改善了传感器的性能,利用最小二乘法线性拟合得到了RCT与Hg2+浓度对数值间呈线性相关,线性范围为0.5 nmol/L~500 nmol/L,检出限为0.2 nmol/L,特异性实验也证明该传感器对Hg2+具有极高的选择性,且有较好的重复性。这一传感器结合了寡核苷酸链生物传感器的高度特异性及电化学分析技术的准确性、便捷性,无需繁复的杂交过程也无需标记电活性基团,减少了操作过程带来的不明确性,避免了电活性基团所带电荷影响反应效果,性能更加稳定,同时起到了简化操作、改善性能和节省成本的作用。
随着纳米电极日渐成熟,本研究有望通过纳米电极来进一步降低检出限。实际水样中往往含有多种重金属离子,这些重金属离子都是对人体健康的巨大威胁,若能同时有效地检测出多种重金属离子,这将是对本项研究实用性的重大提升。此外,在不同形态的汞中,甲基汞毒性最强,但目前测定甲基汞含量的方法主要依赖于大型仪器分析技术,且要与一些样品前处理与分离技术联用,非常繁琐复杂,往后随着甲基汞离子(CH3Hg+)特异性的寡核苷酸链探针的出现与发展,更多的研究将致力于对不同汞形态的选择性测定。
猜你喜欢
化工设计通讯(2022年6期)2023-01-02
北京生物医学工程(2022年4期)2022-08-18
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中国生殖健康(2020年2期)2021-01-18
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
表面工程与再制造(2019年6期)2019-08-24
生物工程学报(2018年5期)2018-06-11
资源节约与环保(2018年1期)2018-02-08
中国医疗保险(2017年5期)2017-05-17
中国康复理论与实践(2015年10期)2015-12-24
