
β-内酰胺酶抑制剂研究进展
2019-07-06 02:18韩江雪刘忆霜肖春玲
中国抗生素杂志 2019年6期
韩江雪 刘忆霜 肖春玲
(中国医学科学院北京协和医学院医药生物技术研究所,北京 100050)
细菌耐药性的产生与传播,使细菌感染性疾病的临床治疗面临巨大的挑战。目前临床上广泛使用的抗细菌药物大多数还是20世纪发现的抗生素或其衍生物,在长期的选择压力下,细菌对已有抗菌药物产生了多种耐药机制,即便曾经被称为“最后一道防线”的碳青霉烯类药物也未能幸免[1]。2016年,我国临床分离的肺炎克雷伯菌对碳青霉烯类的耐药率大于15%,而铜绿假单胞菌和鲍曼不动杆菌对碳青霉烯类的耐药率分别接近30%和70%,均高于2015年的耐药率[2]。
与细菌耐药问题相对应的是新型抗菌药物的匮乏。半个世纪以来,仅有利奈唑胺、达托霉素、贝达喹啉、德拉马尼以及限于局部用药的瑞他帕林5种新型抗菌药物上市[3-5],而这些药物仅对革兰阳性菌和结核分枝杆菌有效,使得革兰阴性菌特别是耐药阴性菌引起的感染成为感染性疾病临床治疗中最大的难题,寻找抗耐药阴性菌的药物迫在眉睫。
针对病原微生物生理代谢筛选和研制药物的进展缓慢。正因为如此,另一种抗耐药细菌药物的研发策略逐渐被重视,即针对耐药机制,筛选和发现抗菌药物的增效剂,瓦解细菌的耐药防线,恢复细菌对临床现有抗菌药物的敏感性。截至目前抗菌药物增效剂中最为成功的就是β-内酰胺酶抑制剂。β-内酰胺类药物具有高效低毒的良好特性,是全球使用最为广泛的一类抗菌药物[6],在我国临床使用率为62.1%。但伴随着β-内酰胺类药物的广泛使用,细菌通过产生β-内酰胺酶、改变细胞壁或外膜的通透性、靶标蛋白突变、以及表达外排泵等机制等对此类药物耐受,而其中最主要的机制是通过产生β-内酰胺酶导致药物水解失活[7],因此筛选新的β-内酰胺酶抑制剂,有可能获得各种新的抗菌药物增效剂,恢复耐药菌对这一大类抗菌药物的敏感性。
1 β-内酰胺酶与细菌耐药性
β-内酰胺酶的作用是对β-内酰胺类药物进行水解使之失活。从化学机制上可以将β-内酰胺酶分为两大类,一类是活性位点为丝氨酸残基的丝氨酸β-内酰胺酶(serine-β-lactamases, SBLs),在Ambler分类上包括A、C、D类β-内酰胺酶,另一类则是活性中心依赖锌离子的金属β-内酰胺酶(metallo-β-lactamases, MBLs),包括B类β-内酰胺酶[8]。在过去的70年间,SBLs,如TEM酶、SHV酶、CTX-M酶、AmpC酶等一直在β-内酰胺类耐药机制中占据主导地位,克拉维酸等SBLs抑制剂在临床上的成功应用也切实改善了β-内酰胺类药物的耐药问题[9]。然而,随着BcII酶、BlaB酶、CcrA酶、IMP-1酶、VIM-2酶、VIM-4酶、VIM-7酶、SPM-1酶和GIM-1酶等MBLs的广泛传播,特别是可水解除氨曲南外几乎所有的β-内酰胺类抗生素的NDM-1的检出,使得β-内酰胺类药物耐药形势更为严峻。近年来,在世界范围内又陆续发现了多种NDM型的β-内酰胺酶(依次命名为NDM-1~NDM-17),这些酶与NDM-1相比有一个或多个位点的氨基酸替换,对β-内酰胺类抗生素的水解活性也有一定的差异[10-11]。
广泛传播的MBLs导致了更为严重的耐药问题:其一,MBLs通常能水解包括碳青霉烯类在内的大多数β-内酰胺类药物,而由于SBLs和MBLs的结构差异,使得现有的SBLs抑制剂(如克拉维酸、舒巴坦等)对MBLs无抑制活性,而且目前临床上尚无可用的MBLs抑制剂;其二,随着SBLs和MBLs共存耐药菌株的增多,即便获得了可用于临床的MBLs抑制剂,也难以单独对抗SBLs和MBLs共存的耐药菌,势必需要更加复杂的联合给药方案[12]。因此,筛选能同时对SBLs和MBLs具有抑制作用的β-内酰胺酶抑制将能更有效地维持临床上最为广泛使用的β-内酰胺类药物的活性。
2 SBLs抑制剂(图1)
至今已有克拉维酸、舒巴坦、三唑巴坦、阿维巴坦和vaborbactam 5种SBLs抑制剂上市,并在临床治疗中应用广泛。SBLs抑制剂与β-内酰胺类药物的联用有效逆转了由于SBLs引起的β-内酰胺类药物耐药问题,此外还有数种不同结构类型的新型SBLs抑制剂已经进入到临床研究阶段[13],期待不远的将来会有多种新结构类型的β-内酰胺类药物增敏剂应用于临床。
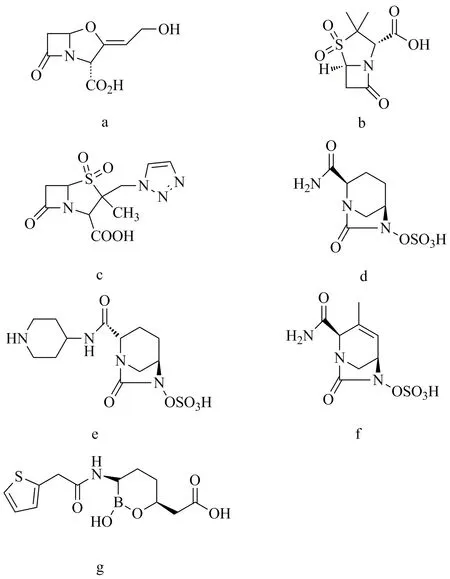
图1 SBLs抑制剂Fig.1 Serine-β-lactamases inhibitors
2.1 具有β-内酰胺结构的SBLs抑制剂
克拉维酸、舒巴坦和三唑巴坦是20世纪70~80年代研制成功的并已广泛应用的SBLs抑制剂,它们均具有β-内酰胺结构,通过与β-内酰胺酶形成稳定而且不可逆的结合,抑制其发挥对β-内酰胺类抗生素的水解作用,从而提高了青霉素类和头孢菌素类抗生素对产SBLs菌株的敏感性[13]。克拉维酸是于1976年从棒状链霉菌的发酵液中分离得到的天然来源的SBLs抑制剂[14],也是第一个用于临床的β-内酰胺酶抑制剂,其结构如图1a。在临床上,常用其合剂克拉维酸/阿莫西林(口服剂)或克拉维酸/替卡西林(注射剂)来治疗由耐药菌导致的院内重度感染。克拉维酸对Richmond type Ⅱ、Ⅲ、Ⅳ和Ⅴ类的β-内酰胺酶有抑制作用[15],对产SBLs的金黄色葡萄球菌、大肠埃希菌、克雷伯菌、变形菌、志贺菌、假单胞菌和流感嗜血菌等都有抑制活性。在低浓度克拉维酸的作用下(<10μg/mL),多种产SBLs的耐药菌对青霉素类和头孢菌素类药物的敏感性都可以恢复到与不产SBLs的细菌相同的水平[14]。
舒巴坦和三唑巴坦[15]是分别于1978年和1984年发现的青霉烷砜类半合成β-内酰胺酶抑制剂,其结构如图1b~c。三唑巴坦与克拉维酸对SBLs的抑制作用很接近,对TEM-1的IC50分别为0.032和0.058μmo/L,对SHV-4的IC50分别为0.055和0.004μmo/L;而舒巴坦的抑制作用比较弱,其对TEM-1和SHV-4的IC50分别为1.56和0.26μmo/L[16]。当32μg/mL舒巴坦与氨苄西林联用时,可将氨苄西林对TEM-31耐药菌的MIC降低16倍;而16μg/mL克拉维酸与氨苄西林联用时,可将氨苄西林对TEM-31耐药菌的MIC降低32倍。这3种抑制剂的抑酶谱及作用机制比较相似,对当时最为常见的SBLs中的A类β-内酰胺酶TEM、SHV具有较强的抑制活性,但对同属SBLs的C类AmpC、D类β-内酰胺酶OXA活性较弱。舒巴坦/氨苄西林合剂、三唑巴坦/哌拉西林合剂均已在美国和欧洲批准上市,三唑巴坦/头孢吡肟合剂在亚洲广泛使用,三唑巴坦/头孢洛扎合剂也分别于2014年和2015年在美国和欧洲批准上市。但是近年来β-内酰胺酶种类逐渐增多,很多的A类β-内酰胺酶对这3种抑制剂敏感性降低,研制新型β-内酰胺酶抑制剂的克服β-内酰胺类药物耐药菌引发的感染任重而道远。
2.2 具有二氮杂环辛烷类化合物(DBO)结构的SBLs抑制剂
新型SBLs抑制剂阿维巴坦于2015年上市,不同于具有β-内酰胺类结构的SBLs抑制剂,是第一个用于临床的二氮杂双环辛烷类化合物(DBO),其结构如图1d。阿维巴坦可以与大多数A类和C类β-内酰胺酶形成可逆的共价键,而其自身结构又可经逆反应得以恢复,因此具有长效抑酶作用[17]。阿维巴坦对TEM-1酶和SHV-4酶的抑制效果强于克拉维酸,其IC50分别为0.008和0.003μmo/L,推测其强活性与其长效抑酶作用有关[16]。Premavathy等[18]利用临床分离的126株铜绿假单胞菌进行阿维巴坦与头孢他啶联用的体外抗菌实验,当4μg/mL阿维巴坦分别与头孢他啶、氨曲南、亚胺培南、哌拉西林联用时,均可在不同程度上防止β-内酰胺类药物被水解。其中,单独使用头孢他啶时,有65%的分离株对其敏感,而4μg/mL阿维巴坦与头孢他啶联用时,有94%的分离株对其敏感。在当前的临床治疗中,阿维巴坦常与头孢他啶联用,阿维巴坦/头孢他啶合剂分别于2015年和2016年在美国和欧洲批准上市,而阿维巴坦/氨曲南合剂、阿维巴坦/头孢洛林合剂仍在进行临床二期试验。
与阿维巴坦结构相似的relebactam是针对A类和C类β-内酰胺酶设计的含有哌啶基的二氮杂双环辛烷(DBO)类抑制剂,其结构如图1e。其作用机制与阿维巴坦相似。有研究结果表明其对铜绿假单胞菌中的AmpC酶、KPC-2酶以及鲍曼不动杆菌中的AmpC酶的IC50分别为410、210和4100nmol/L,从酶抑制活性上看弱于阿维巴坦。当亚胺培南与4μg/mL relebactam联用时,可将亚胺培南对AmpC耐药菌的MIC降低4倍,将亚胺培南对含KPC的肺炎克雷伯菌和肠杆菌科细菌的MIC从16~64μg/mL降至0.12~1μg/mL[19]。但是relebactam对D类碳青霉烯酶OXA-40(OXA-24)酶没有抑制作用,它只能逆转产A类和C类SBLs的耐药菌对β-内酰胺类药物耐药性,对产D类碳青霉烯酶的耐药菌无能为力。目前,relebactam/亚胺培南/西司他丁已完成复杂腹腔内感染的临床二期试验,复杂尿路感染临床三期试验仍在进行中。
另外一个与阿维巴坦结构相似的二氮杂环辛烷类化合物结构的ETX2514是由阿斯利康合成的SBLs抑制剂,目前舒巴坦- ETX2514合剂已进入二期临床研究,其结构如图1f。ETX2514对A、C、D类SBLs均有很好的抑制活性,对KPC-2、AmpC及OXA-24的IC50值分别为0.004、0.014和0.19μmol/L[20]。在体外抗菌实验中,4μg/mL ETX2514与哌拉西林联用时,可将哌拉西林对KPC-2耐药菌、AmpC耐药菌及OXA-24耐药菌的MIC分别降低16、32和8倍以上[21]。此外ETX2514在分别与哌拉西林、美罗培南、舒巴坦联用时,提高对含A、C、D类SBLs鲍曼不动杆菌耐药菌对β-内酰胺类药物敏感性的能力均强于阿维巴坦,表现出更好的抑制活性[20]。
2.3 非β-内酰胺类结构的有机硼酸类SBLs抑制剂
Vaborbactam(RPX7009)是于2017年上市的有机硼酸类SBLs抑制剂,其结构如图1g。Vaborbactam通过与丝氨酸蛋白酶之间发生相互作用而发挥抑酶作用,vaborbactam对多种A类(KPC-2、CTX-M-15、 SHV-12、TEM-10)和C类(P99)的β-内酰胺酶具有抑制作用,其Ki值分别为0.069、0.044、0.029、0.110、0.053和0.099μmol/L。除此之外,对部分D类β-内酰胺酶也有显著的抑制活性[22]。Vaborbactam(RPX7009) 是一种在体内外均具有广谱抑制活性的SBLs抑制剂,4μg/mL的vaborbactam与美罗培南联用,可以将美罗培南对多种含A类β-内酰胺酶(KPC-2/KPC-3、SME和NMC-A)的耐药菌的MIC降低至空载体对照菌的水平;联用其他的β-内酰胺类抗生素,也可有效治疗多重耐药革兰阴性菌引发的感染[23]。其作用机制的研究表明,vaborbactam通过与丝氨酸蛋白酶之间发生相互作用而发挥了抑酶作用,但是vaborbactam对11种不同的哺乳动物丝氨酸蛋白酶的IC50值均大于1000μmol/L,表明其对SBLs的抑制作用具有特异性。目前,vaborbactam/美罗培南合剂已于2017年在美国批准上市。
3 MBLs抑制剂
虽然已经上市的以及处于研究阶段的SBLs抑制剂均对SBLs表现出较强的抑制活性,但是对MBLs却没有抑制作用,不能有效解决因产生MBLs而导致的β-内酰胺类抗生素耐药问题,MBLs抑制剂的研发迫在眉睫。
目前文献报道的MBLs抑制剂的抑制机制主要分为两类,第一类是作用于MBLs活性中心的锌离子:其中一种为金属离子螯合剂,通过螯合MBLs活性中心的锌离子,从而抑制MBLs的水解活性,如来源于真菌代谢产物的aspergillomarasmine A(AMA)、金属离子螯合剂1,4,7-三氮杂环壬烷-1,4,7-三乙酸(NOTA)和1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(DOTA);另一种为带有二羧基的酸,如马来酸、琥珀酸、邻苯二甲酸等,通过与MBLs活性中心的锌离子结合,从而抑制MBLs的水解活性;第二类抑制剂如ebselen、magnolol等,则是作用于MBLs的氨基酸残基。
3.1 Aspergillomarasmine A(AMA)
Aspergillomarasmine A(AMA)是一种来源于真菌发酵液的天然产物,它是由King等于2014年已经通过细胞水平的筛选得到的MBLs抑制剂,是作用于活性中心锌离子的MBLs抑制剂,AMA结构如图2a。AMA结构上与乙二胺四乙酸相似的锌离子螯合剂,但与乙二胺四乙酸相比,AMA的毒性更低且耐受性更好,说明AMA可能具有较好的研发前景。AMA对MBLs的抑制作用具有选择性,它能够快速有效的抑制NDM-1酶和VIM-2酶,对NDM-1酶和VIM-2酶的IC50值分别为(4.0±1.0)和(9.6±2.4)μmol/L,Ki分别为11和7nmol/L,但是对IMP-7的抑制作用很弱;对TEM-1和CTX-M-15两种SBLs没有表现出抑制活性。体外的药效学研究表明:美罗培南与2μg/mL浓度的AMA联合使用,可有效杀灭含有VIM或NDM型等位基因的肠杆菌科细菌、不动杆菌和假单胞菌等对美罗培南耐药的菌株,几乎完全恢复了这些耐药微生物对美罗培南的敏感性;体内的药效学研究结果显示:在感染了表达NDM-1的肺炎克雷伯菌的小鼠中,10mg/kg美罗培南与30mg/kg AMA单剂量联用可以使感染小鼠的存活率大于95%,有效地恢复了美罗培南的活性,表明AMA和碳青霉烯类抗生素的组合具有治疗产MBLs碳青霉烯耐药病原体的潜力[24-25]。
3.2 NOTA和DOTA

图2 MBLs抑制剂Fig.2 Metallo-β-lactamases inhibitors
1,4,7-三氮杂环壬烷-1,4,7-三乙酸(NOTA)和1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(DOTA)是Somboro于2015年发现的金属离子螯合剂类型的MBLs抑制剂,其结构如图2b~c。NOTA和DOTA可有效地抑制NDM-1、NDM-4、VIM-1、IMP-1、IMP-8的活性,二者对MBLs的抑制作用强于AMA,在低至0.06mg/L浓度下,NOTA与DOTA仍可恢复碳青霉烯类抗生素对MBLs产生菌的抑制活性;但在与美罗培南联用时二者对耐药菌抑制活性显示出差异:美罗培南/NOTA组合的抗耐药菌活性比美罗培南/DOTA组合的抗耐药菌活性高32倍。这类金属螯合剂联合使用亚胺培南时也可有效地恢复亚胺培南对产MBLs耐药菌株的活性,表明这类抑制剂恢复碳青霉烯类类药物对耐药菌活性的作用具有普遍性。对NOTA和DOTA的细胞毒性研究发现,这些化合物在应用浓度下没有出现溶血反应,这一结果表明其他的锌离子螯合剂也有可能作为MBLs抑制剂应用于临床的抗耐药菌治疗[26],但金属离子螯合剂对哺乳动物体内含锌酶(如血管紧张素转化酶、乙醇脱氢酶及哺乳动物羧肽酶等)可能存在的抑制活性,是上述MBLs抑制剂在进行药物研发时必须面对的风险。
3.3 ME1071
ME1071是由Meiji Seika Kaisha Ltd公司开发的一种马来酸衍生物,其结构如图2d。ME1071对IMP-1及VIM-2具有明确的抑制活性,Ki值分别为0.41和120μmol/L[27]。当ME1071为32μg/mL时,可显著提高产NDM-1、IMP-1及VIM-2的耐药菌对美罗培南、亚胺培南、比阿培南、多利培南这4种碳青霉烯类抗生素的敏感性,其中ME1071对NDM-1耐药菌耐药性的逆转效果弱于产IMP-1及VIM-2的耐药菌,推测这与ME1071对NDM-1的亲和力弱于IMP-1及VIM-2有关[28]。对ME1071作用机制研究结果表明,该化合物是通过与MBLs活性中心的锌离子结合,从而抑制了MBLs对β-内酰胺类药物的水解活性。目前对ME1071的研究已经完成了Ⅰ期临床实验,期待其后续的研究进展。
3.4 Ebselen
Ebselen是Chiou等[29]在2015年通过以含有pET28b-blaNDM-1质粒的E.coli BL21为模式生物的全细胞模型筛选得到的非锌离子依赖的MBLs抑制剂,其结构如图2e。Ebselen对NDM-1有明确的抑制活性,Ki值为(0.38±0.03)μmol/L。体外研究发现:Ebselen可以逆转产NDM-1的耐药菌对β-内酰胺类药物的耐药性,当Ebselen与美罗培南和氨苄西林分别以摩尔比1.3:1和1.4:1联用时,耐药菌对二者的敏感性分别提高128倍和16倍。对Ebselen作用机制的研究结果显示:该化合物与NDM-1活性位点的Cys221形成S-Se键,二者之间具有强亲和力,并可以从活性位点移除氢离子,使NDM-1处于失活状态下,进而无法发挥水解活性。Ebselen有可能发展成为作用于B1和B2类MBLs的广谱β-内酰胺酶抑制剂,然而,其抗氧化活性和毒性也限制了此类药物的开发。
3.5 Magnolol
Magnolol(木兰醇)是Liu等[30]于2018年通过NDM-1抑制剂模型从木兰树皮分离的天然产物中筛选得到的NDM-1抑制剂,其结构如图2f,Magnolol可有效地抑制NDM-1的活性,其IC50为6.47μg/mL。在体外抗菌实验中,联用32μg/mL Magnolol可以将美罗培南对产NDM耐药菌株E.coli ZC-YN3(NDM-1)、E.coli ZC-YN5(NDM-5)、E.coli ZC-YN7(NDM-9)的MIC降低4倍,且在3h内杀死几乎所有细菌,显示出具有开发成为MBLs抑制剂的潜力。其作用机制研究发现:Magnolol可通过直接与NDM-1的110-200残基附近的催化口袋结合,且与Ser217形成氢键,抑制NDM-1的活性。此外还有5个氨基酸残基(Val73、Lys211、Leu218、Gly219、His250)与Magnolol的结合相关。
4 SBLs/MBLs双重抑制剂
SBLs和MBLs在结构上差异明显,通常一种酶抑制剂都特异性地抑制一种β-内酰胺酶,但在抑制剂筛选过程中仍发现了对两类β-内酰胺酶均表现出抑制活性的化合物。
Schofield等[31]通过化学合成得到了环硼酸酯类化合物,该类化合物对SBLs/MBLs具有双重的抑制活性,而环硼酸酯类化合物2为Schofield等设计合成的5个化合物中活性最好的一个化合物,其结构如图3。环硼酸酯类化合物2对SBLs和MBLs均有明显的抑制活性,其对TEM-1和VIM-2、NDM-1的IC50分别为0.003、0.003和0.029μmol/L。在体外抗菌实验中,25μg/mL环硼酸酯类化合物2与美罗培南联用时,可将美罗培南对NDM-1耐药菌的MIC降低32倍。对其作用机制的研究表明:该化合物通过模拟β-内酰胺类药物被酶水解的过程中产生的高能四面体中间产物,发挥抑制SBLs和MBLs活性的作用。这类化合物的发现为研发SBLs/MBLs双重抑制剂的可行性提供了支持。有研究发现环状硼酸盐也可以通过相同的机制有效地抑制一些青霉素结合蛋白(PBP)的活性。该研究结果提示我们,通过筛选模拟常见的高能四面体中间化合物有可能获得具有MBLs、SBLs或PBP抑制活性的新型β-内酰胺类抗生素增效剂。
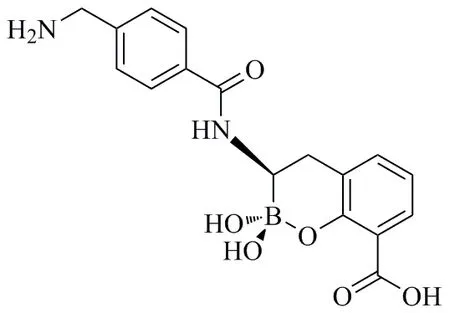
图3 SBLs/MBLs双重抑制剂Fig.3 Dual action inhibitors of SBLs and MBLs
单一类别的化合物能够同时抑制SBLs和MBLs的实验结果在本实验室的药物筛选中也被发现了,通过联合使用SBLs和MBLs抑制剂筛选模型,本研究筛选得到了化合物IMB-XW1和IMB-XH5,这两个化合物对A类SBLs中的BlaC和MBLs中的NDM-1都具有抑制活性,IMB-XW1对BlaC和NDM-1的IC50分别为9.75和1.49μmol/L,IMB-XH5对BlaC和NDM-1的IC50分别为2.24和0.25μmol/L。由此我们相信,联合使用SBLs和MBLs抑制剂高通量筛选模型,从不同来源的样品中筛选SBLs/MBLs双重抑制剂,有可能获得新型的β-内酰胺酶抑制剂,从而获得可有效治疗具有多重耐药机制的耐药菌引发的感染,为耐药阴性菌的临床治疗提供有效的药物。
5 结语
随着各种耐药菌的不断出现和广泛传播,细菌耐药问题成为世界范围内威胁公众健康的重要问题。β-内酰胺类抗生素是目前应用最为广泛的抗生素,但陆续出现的多种耐药机制,大大降低了β-内酰胺类抗生素的治疗效果。针对其耐药机制,研制新型的β-内酰胺酶抑制剂,是逆转此类药物耐药性的重要手段,而研究人员在SBLs抑制剂、MBLs抑制剂、以及SBLs/MBLs双重抑制剂的研究方面所取得的进展,为获得新型β-内酰胺类药物的增效剂带来了希望,虽然距离将其开发成临床可用的药物还需要艰难而漫长的历程,但我们坚信在药学工作者的不断努力下,新型β-内酰胺类药物增效剂会在不久的将来应用于耐药菌的临床治疗。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
当代医药论丛(2022年21期)2022-12-21
中国感染与化疗杂志(2022年4期)2022-12-11
中国现代中药(2022年4期)2022-05-08
食品安全导刊(2021年34期)2021-11-28
中华养生保健(2020年1期)2020-11-16
中国食品(2020年18期)2020-10-15
中国药业(2019年3期)2019-01-26
科学与财富(2017年22期)2017-09-10
赤子(2017年4期)2017-06-30
