
Identification of anticancer drugs to radiosensitise BRAFwild-type and mutant colorectal cancer
2019-06-18 07:06RebeccaCarterAzadehCheraghchiBashiAdamWesthorpeShengYuYasminShanneikElenaSeraiaDjamilaOuaretYasuhiroInoueCatherineKochJennyWildingDanielEbnerAndersonRyanFrancescaBuffaRickySharma
Cancer Biology & Medicine 2019年2期
Rebecca Carter, Azadeh Cheraghchi-Bashi, Adam Westhorpe, Sheng Yu, Yasmin Shanneik, Elena Seraia,Djamila Ouaret, Yasuhiro Inoue, Catherine Koch, Jenny Wilding, Daniel Ebner, Anderson J. Ryan, Francesca M. Buffa, Ricky A. Sharma
1NIHR University College London Hospitals Biomedical Research Centre, UCL Cancer Institute, University College London,London WC1E 6DD, UK;
2NIHR Oxford Biomedical Research Centre, Department of Oncology, University of Oxford, Oxford OX1 2JD, UK;
3Computational Biology and Integrative Genomics, University of Oxford, Oxford OX1 2JD, UK;
4NDM Research Building, Nuffield Department of Medicine, University of Oxford, Oxford OX1 2JD, UK;
5Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, University of Oxford, Oxford OX1 2JD, UK;
6Mie University, Graduate School of Medicine,Department of Gastrointestinal and Pediatric Surgery, Division of Reparative Medicine, Institute of Life Sciences, Edobashi 2-174, Tsu, Japan;
7Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02142, USA;
8Target Discovery Institute, National Phenotypic Screening Centre, Nuffield Department of Medicine, University of Oxford,Oxford OX1 2JD, UK;
9CRUK & MRC Oxford Institute for Radiation Oncology, University of Oxford, Oxford OX1 2JD, UK
ABSTRACT Objective:Patients with BRAF-mutant colorectal cancer (CRC) have a poor prognosis. Molecular status is not currently used to select which drug to use in combination with radiotherapy. Our aim was to identify drugs that radiosensitise CRC cells with known BRAF status.Methods:We screened 298 oncological drugs with and without ionising radiation in colorectal cancer cells isogenic for BRAF. Hits from rank product analysis were validated in a 16-cell line panel of human CRC cell lines, using clonogenic survival assays and xenograft models in vivo.Results:Most consistently identified hits were drugs targeting cell growth/proliferation or DNA damage repair. The most effective class of drugs that radiosensitised wild-type and mutant cell lines was PARP inhibitors. In clonogenic survival assays, talazoparib produced a radiation enhancement ratio of 1.9 in DLD1 (BRAF-wildtype) cells and 1.8 in RKO (BRAF V600E) cells. In DLD1 xenografts, talazoparib significantly increased the inhibitory effect of radiation on tumour growth (P ≤ 0.01).Conclusions:Our method for screening large drug libraries for radiosensitisation has identified PARP inhibitors as promising radiosensitisers of colorectal cancer cells with wild-type and mutant BRAF backgrounds.
KEYWORDS Radiosensitizer; colorectal cancer; PARP inhibitor; radiotherapy
Introduction
Colorectal cancer (CRC) is one of the most common forms of cancer, accounting for approximately 1 in 10 new cancer diagnoses worldwide in 20121. Radiotherapy is commonly used to treat rectal cancers prior to surgery or to treat inoperable colorectal metastases, in the form of stereotactic body radiotherapy or selective internal radiotherapy2-4.
International standard combination therapy for rectal cancer, radiotherapy delivered with 5-fluorouracil (5FU) as a radiosensitiser, is given either as an infusion or as an oral prodrug (capecitabine). There is currently no molecular basis for the selection of patients for radiotherapy, nor for the selection of any alternative drug to use as a radiosensitiser.With the current standard, sufficient downsizing by chemoradiotherapy is obtained by approximately half of patients treated5. There is scope for improving the radiotherapy approaches currently offered to patients.Clinical trials have added additional drugs to 5FU as a combination radiosensitising approach6,7without molecular selection, but these trials have not changed the international standard.
Colorectal tumours have a heterogeneous molecular background8. Commonly occurring CRC mutations that may be prognostic or can affect treatment decisions include KRAS, BRAF and PIK3CA mutations, which are found in 42%, 9% and 13% of CRC patients respectively9. KRAS,BRAF and PIK3CA are vital components of two main cellular signalling pathways; RAS/MEK/ERK and PI3K/AKT/mTOR;strongly inter-connected pathways that play central roles in tumorigenesis by regulating cell survival, proliferation,metabolism, and motility. The KRAS gene is a member of the oncogenic RAS gene family and binds to effector kinases including BRAF and phosphatidylinositol 3-kinase (PI3K).The PIK3CA gene encodes the PI3K p110α subunit, which interacts with RAS proteins10.
The commonest BRAF mutation in colorectal cancer, the V600E substitution, results in elevated kinase activity and constitutive downstream MEK and ERK phosphorylation11,12. The presence of BRAF V600E in advanced CRC correlates with poor prognosis with markedly worse progression after chemotherapy13-15. BRAF mutation is predictive of poor response to cetuximab in metastatic CRC,also observed for KRAS and PIK3CA mutations16-18.Although patients with BRAF-mutant cancers do less well with chemotherapy, anti-EGFR therapies and surgery19, there is currently no suggestion that they benefit less from radiotherapy. Although BRAF mutation is relatively rare in rectal cancer, radiotherapy can also be used to treat inoperable liver metastases from CRC. It has been suggested that CRC liver metastases respond less well to radiotherapy than liver metastases from other primary malignancies20,hence the addition of a radiosensitising drug may be of value to improve the therapeutic index during radiotherapy21.
Our aim was to develop a radiosensitiser drug discovery assay enabling identification of drugs that will enhance radiotherapy more effectively than the current standard,5FU, and demonstrate activity in defined molecular backgrounds. Firstly, we developed a high throughput screen(HTS), in CRC cell lines, to identify drugs that could be effective radiosensitisers in the context of BRAF V600E activating mutations. The drugs identified during the screen were validated across an extensive panel of human CRC cell lines, selected to represent aspects of the molecular landscape of CRC; including BRAF V600E in both MSI and MSS backgrounds, and a spectrum of KRAS, PIK3CA and p53 mutations. Such cell line panels recapitulate the different subtypes found in CRC, are representative of genetic alterations found in primary cancers and are good predictors of clinical efficacy during drug development programmes22.Here, we use this model to test new drug-radiotherapy combinations for the first time, identifying PARP inhibitors as the most strongly radiosensitising class of agent before validating by clonogenic survival assays and in vivo xenograft studies.
Materials and methods
Cell lines, drug library and irradiations
The parental CRC cell lines RKO (BRAF V600E/V600E/WT)and VACO432 (V600E/WT) and their isogenic pairs RKOT29 (BRAF WT/-/-) and VACO432-VT1 (BRAF WT/-) were a gift from Sandra Van Schaeybroeck, Queens University,Belfast, UK (mutation status confirmed by sequencing). The panel of colorectal cancer cell lines utilised for cell proliferation assays was obtained from Prof. Walter Bodmer,University of Oxford, UK. The cell line panel is listed in Supplementary Table S1, and has been previously described22. Non-malignant cell lines were obtained from Prof. Gillies McKenna, University of Oxford, UK. All cell lines were used within 12 passages, or where necessary,replenished using frozen aliquots of the initial passage.Isogenic cell lines were grown in McCoy's 5A (Modified)Medium, and other cell lines in DMEM; both supplemented with 10% Fetal Bovine Serum and 1 × penicillin/streptomycin(Thermofisher Scientific Inc., MA, USA), in a 37°C, 5% CO2,humidified incubator. The small compound anti-cancer drug library was provided in 384-well plate format (Target Discovery Institute, University of Oxford), and contained 222 drugs from the TDI Extended Oncology Drugs Library(ODL) and 76 from the NCI Developmental Therapeutics Program (DTP) Approved Oncology Drug set(Supplementary Table S2).
A GSR D1 irradiator (Gamma-Service Medical GmbH,Leipzig, Germany) a Cs-137 source, (dose rate 1.5 Gy/min)was used for cell irradiations. For xenografts, a RS320 X-ray irradiator (Gulmay Limited, Byfleet, UK) was used (1.6 Gy/min), with lead shielding to localise dose to tumor.Dosimetry was calculated from optical density of scanned Gafchromic EBT3 film (Ashland, NJ, USA), corrected and calibrated to the National Physical Laboratory (Teddington,UK) primary standard.
High-throughput drug screen with ionising radiation
Methodology and data analysis followed internationally recognised high-throughput screening guidelines23. BRAF V600E isogenic RKO and VACO432 cells were seeded in 52 μL/well by Flexdrop (PerkinElmer, MA, USA). Seeding density in 384-well plates was 300 cells/well (RKO) and 1,000 cells/well (VACO432). Eighteen hours after seeding, cells were screened with 298 oncological drugs, in 5-fold dilutions from 10 μM-16 nM. Janus workstations (PerkinElmer, MA,USA) were used to transfer 13 μL of compound from library plate to cell culture plates. Positive controls were PI103 and vorinostat, negative controls were vehicle (DMSO) alone.After 6 h, plates were either mock-irradiated, or irradiated with 4 Gy. Media was replaced 24 h following treatment, and surviving cells allowed to proliferate for five doubling times as optimised in preliminary screens. Cell viability was measured by resazurin (10 μg/mL) in phenol red-free DMEM. Metabolically viable cells reduce resazurin to fluorescent resorufin, which was quantified by PerkinElmer Envision microplate reader (540 nm excitation/590 nm emission). Control wells reached 90%-100% confluency at the time of assay performance, control irradiated wells were around 60% confluent. Raw data were normalized by rescaling to plate mean intensity and to negative controls.Quality plots were contrasted to assess artifacts and reproducibility. Normalized data Z are presented, as the applied rescaling by plate mean is effectively a z-score standardization. Selection of candidate hits was based on rank product analysis, adapting a published method24.Specifically, for each pair of conditions (i.e. with/without irradiation), the differences between normalised screen intensities were calculated for each well, hence each drug.These differences are presented as Delta-Z (ΔZ) scores. Rank product applied to these differences identified compounds producing large and consistent changes. Probability of false discovery was computed by permutation, with n = 100.Analyses were implemented in R version 2.1 (https://cran.rproject.org/); heatmaps were generated by modifying D3.js libraries (https://d3js.org/).
Cell proliferation and colony formation assays
Our method for comparison of IC50in the presence or absence of radiation has been described previously25.Clonogenic survival was measured following a standard method, with plating efficiency and surviving fractions calculated as described26. Briefly, cells were seeded into 10 cm culture dishes, normally 500 cells/plate (for 0 Gy plates),increasing by 10-fold for each 4 Gy administered, to 500,000 cells/plate (12 Gy). After attachment (overnight), cells were drug-treated, and six hours later exposed to 0, 4, 8 or 12 Gy radiation. Culture medium was replaced 24 hours postirradiation, plates were incubated to form visible colonies >50 cells (10 - 15 days) and fixed with 0.4% methylene blue in methanol. Survival curves were fitted using Graphpad Prism v7.0A. Radiation enhancement ratio (RER) was obtained from the ratio of radiation dose at 1% survival of vehicle compared with drug treated cells.
Xenograft studies
Animal experiments were performed following local ethical review under licence from the UK Home Office (ASPA 1986,revised January 2013). Female Balb/c nude mice (6-8 weeks old) were anaesthetised with 2% isoflurane and subcutaneously injected with 50% matrigel containing 5x106DLD1 cells or 5 × 106RKO/mouse (n = 24) into the back.When tumor volume reached 100 mm3, mice were randomly placed into 4 groups (n = 6/group). Oral treatments were by gavage, in two doses on the first and fourth days of treatment. Group (1) received vehicle only, 10%dimethylacetamide/6% solutol HS/PBS (0.1mL/10 g body weight). Group (2) received talazoparib; 0.1 mg/kg in vehicle.Radiation treatments comprised 2 × 5 Gy, localised to the tumor, also on the first and fourth days of treatment. Group(3) received radiation only, 5 Gy one hour after each vehicle treatment. Group (4) received combination treatment, 5 Gy one hour after each talazoparib treatment. Tumor size was measured by caliper 3 × per week. Mice were sacrificed when tumours reached 400 mm3or 42 days following the first treatment. Tumours were formalin fixed and stained for the hypoxia marker CA9 as previously described27.
Results
Development of a high throughput screen with ionising radiation
In order to identify drugs that radiosensitise CRC cells mutated for BRAF V600E, isogenic cell lines containing either BRAF V600E or BRAF WT variants were screened against a 298-compound library of approved anticancer drugs. Mutation status for KRAS, PIK3CA and p53 for these cell lines is shown in Figure 1A, with the screen protocol outlined in Figure 1B.
A prerequisite for high-throughput detection of radiosensitisers is an assay that is predictive of the effects of drug/ radiation combinations on clonogenic cell survival.Extended incubation following irradiation improves correlation with radiosensitisation28, and we incorporated 5 days incubation following radiation treatment; improving correlation to clonogenic survival, but avoiding compromises to cell metabolism and thus assay performance29. Serial dilution of cells in the presence of resazurin showed equivalent fluorescence, linear in relation to cell number, for both non-irradiated cells, and cells 5 days post-irradiation(data not shown). This indicates that the metabolic assay was a good surrogate for cell number at this timepoint.
Screens were carried out in duplicate and quality plots demonstrated good reproducibility (Figure 1C), with mean Pearson correlation between pairs of replicates of 0.88 and average Z factor of 0.58 for irradiated and 0.53 for nonirradiated plates. Cell viability was compared between normalized irradiated and non-irradiated plates, generating heatmaps of the difference, ΔZ, for each compound. Hit selection (Figure 1D) was based on rank product analysis,with the probability of false discovery computed by permutations (see Materials and methods). Potential hits were drugs that sensitised the BRAF-mutant isogenic variant,at one or more concentrations, with probability of false positive (PFP) ≤ 0.05. Some plates showed a pronounced‘edge effect’, and for this reason, analysis was repeated considering the edge wells as a separate population (Figure 1E). Hits with significant ΔZ score between irradiated and non-irradiated samples, with radiosensitisation factor < 1(normalised against control plates) and P-value ≤ 0.05 were selected as significant. Positive controls were consistently identified as hits, with ΔZ scores ≤ 2, comparable to results obtained in manual assays.
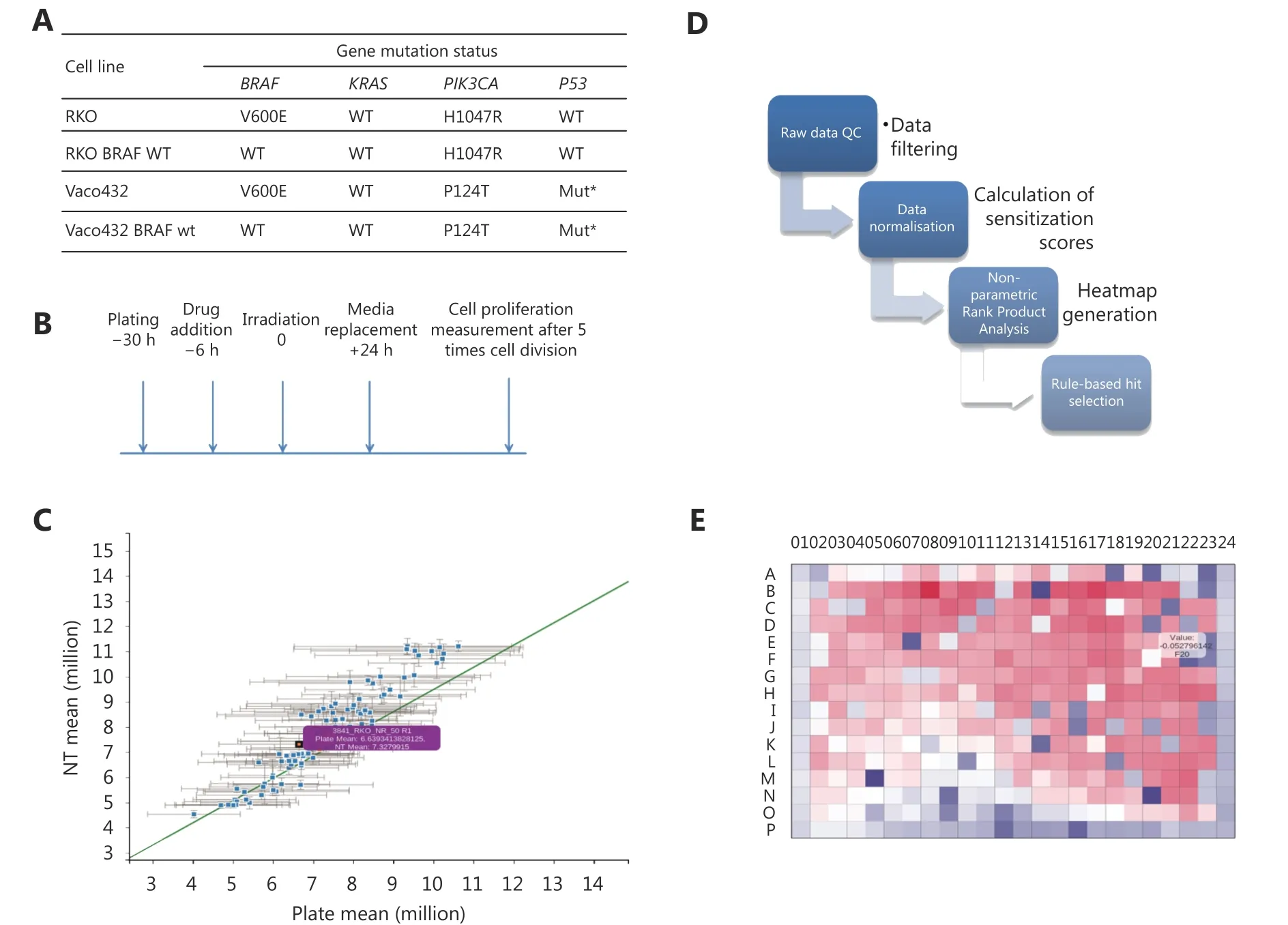
Figure 1 High-throughput screening of FDA approved cancer drugs to identify which drugs should be used for radiosensitisation in the context of single gene mutations in colorectal cancer. (A, B) CRC cells isogenic for BRAF V600E and with defined KRAS, PIK3CA and p53 status were screened with the DTP approved oncology drug library +/- irradiation and allowed to grow for five doubling times. Cell viability was compared between irradiated and non-irradiated plates. (C) Raw data were normalized by rescaling both to the plate mean and negative controls, and quality plots contrasted. (D) Heatmaps were generated for each individual plate. ΔZ scores were calculated between irradiated and non-irradiated plates. Selection of candidate hits was based on a rank product method (see methods). Probability of false discovery was computed by permutation, with 100 permutations. (E) Example heatmap generated for one of the HTS plates. Hits were identified as drugs with a ΔZ score significantly higher than expected by chance when irradiated and non-irradiated samples were compared.
BRAF V600E screen in isogenic cell lines following irradiation
Drugs were ranked according to radiosensitisation against BRAF-mutated cells. The fifteen drugs with the highest significance against BRAF-mutated cells are shown in Table 1.Seven hits have previously been identified as radiosensitisers in the published literature30-36, helping to validate our methodology. Five hits were inhibitors of RAS/RAF/MEK/ERK pathway (trametinib, TAK-733, pimasertib,doramapoimod and dactolisib), predominantly acting in BRAF WT and V600E. Eight drugs reached significance in the BRAF-mutant cell line but not in BRAF WT, including the CHK1 inhibitor, PF477736. Another CHK1 inhibitor,AZD7762, radiosensitised both BRAF variants.
The poly(ADP-ribose) polymerase (PARP) inhibitor,olaparib, significantly increased sensitivity to irradiation in BRAF V600E RKO cells. In a separate screen of BRAF isogenic Vaco432 cells, olaparib also radiosensitised BRAF V600E Vaco432 cells at 16 nM and 80 nM (P ≤ 0.05, data not shown). Based on these data, radiosensitisation by PARP inhibitors (PARPi) in RKO isogenic for V600E and WT, was validated by long-term proliferation assay at a broad concentration range and by clonogenic cell survival assay(Figure 2). Olaparib as a single agent had little effect on survival, but combination treatment caused a significant increase in radiation sensitivity, albeit with similar effect in both BRAF WT and V600E variants.
Radiosensitisation in an extended CRC cell line panel
To validate the screen, we used a cell line panel inclusive of the different molecular subtypes of CRC. We specifically prioritised the drug hits with the most immediate scope for translation to clinical trials in combination with radiotherapy. The cell line panel was selected so that several cell lines exhibited each gene mutation of interest. Fifteen cell lines with defined BRAF, p53, KRAS, PIK3CA and mismatch repair status were used. The compounds chosen for further testing are shown in Table 2, along with p-values indicating whether significant IC50shift was observed following normalisation for radiation effect. The complete IC50results determined by these assays are shown in Supplementary Table S3.
From these assays, olaparib and rucaparib displayed potentradiosensitising ability across multiple cell lines. IC50curves(normalised for radiation effect) were significantly different(P ≤ 0.01) for all except three cell lines; namely, C10, CW2 and Colo678 (Table 2).
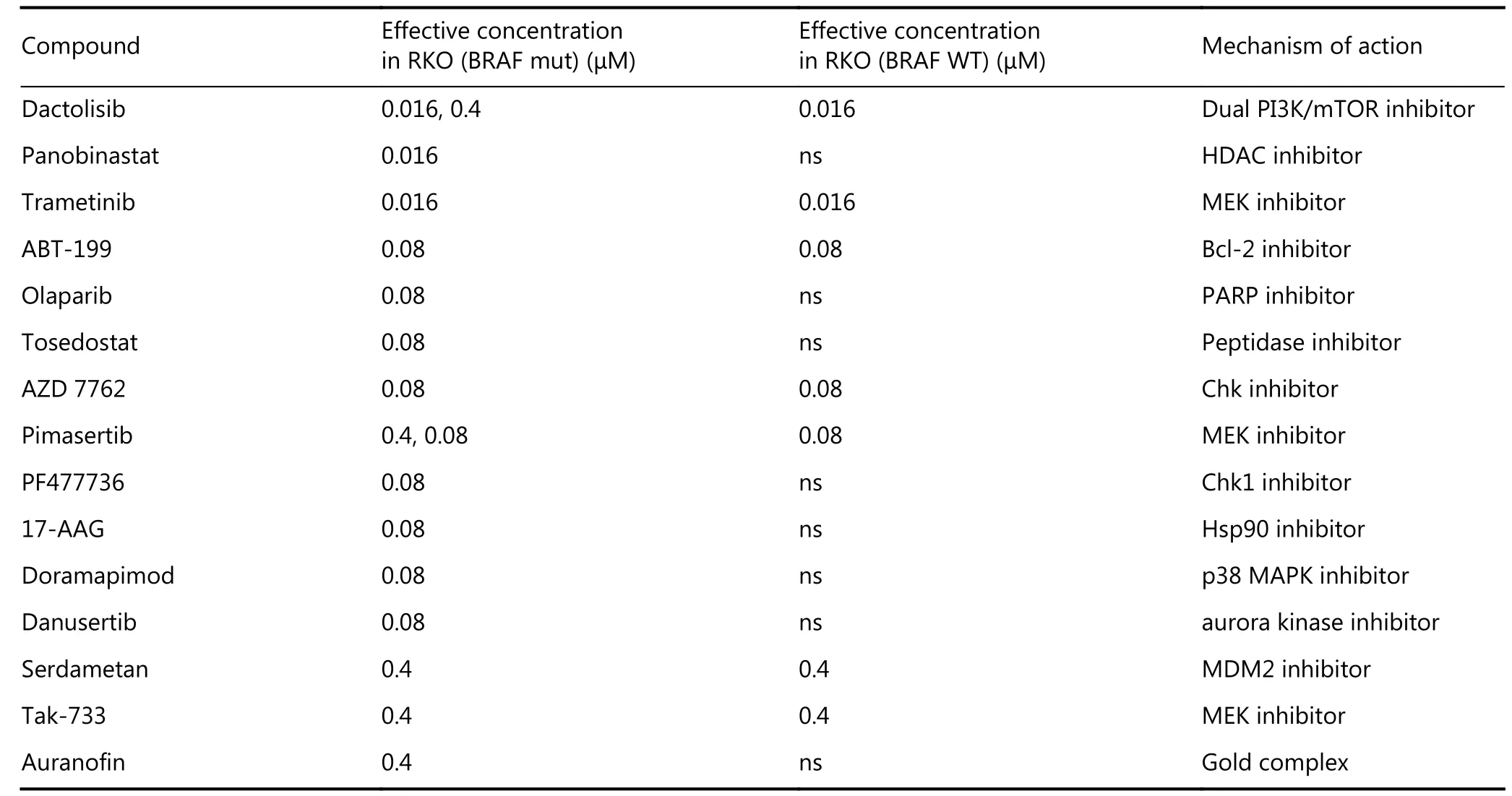
Table 1 Fifteen radiosensitisers identified for BRAF-mutant cells
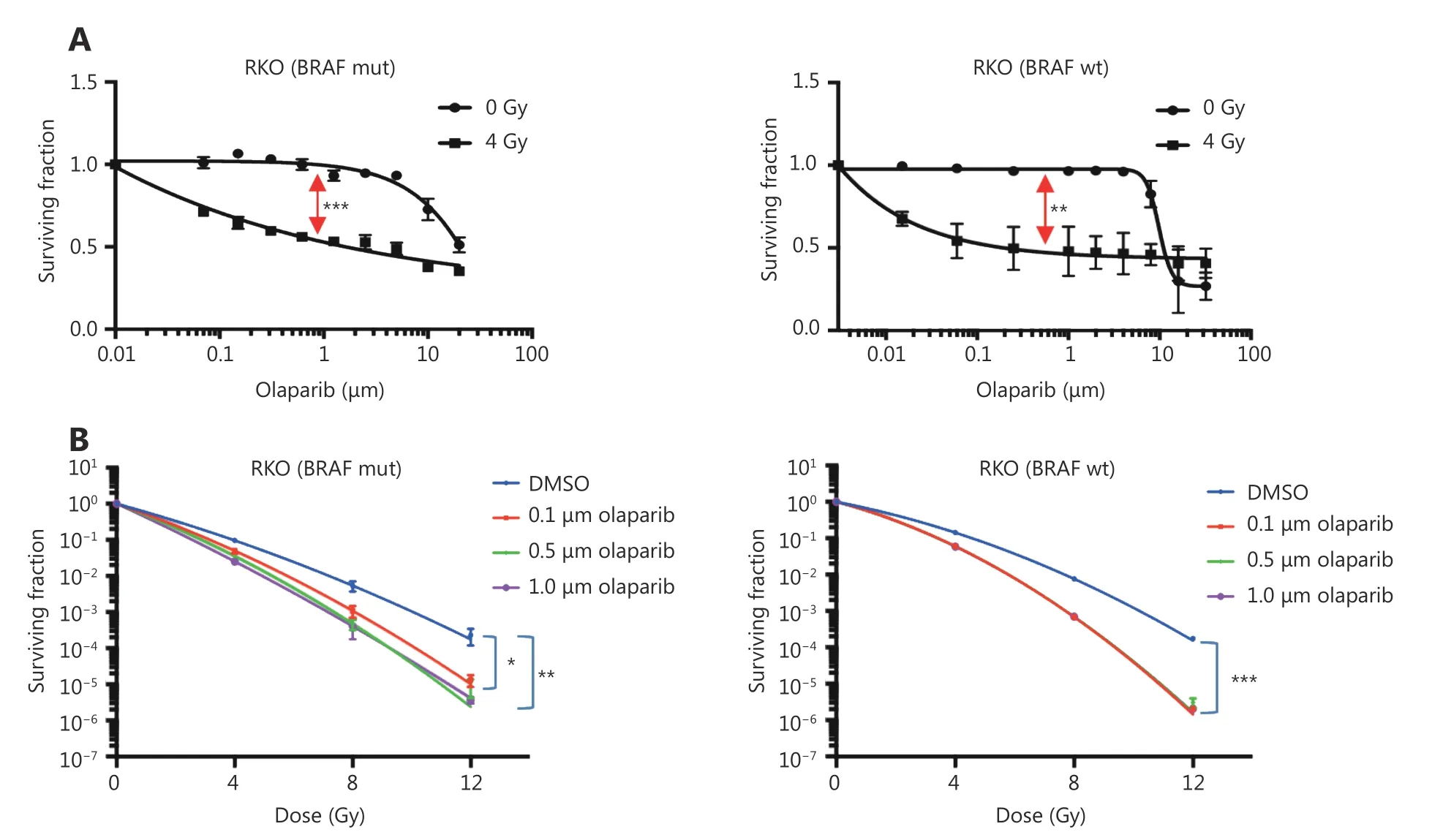
Figure 2 Validation of radiosensitisation effects of olaparib in BRAF-mutant and BRAF-WT isogenic CRC cells. Confirmation of radiosensitisation by olaparib in BRAF mutant and WT RKO cells by: (A) Long-term cell proliferation assays, showing separation (red arrows)between IC50 curves normalised for radiation effect, indicating significant radiosensitisation (BRAF-mutant: P ≤ 0.001; BRAF-WT: P ≤ 0.01,calculated by paired t-test). (B) Clonogenic survival assays, showing significant radiation enhancement by 0.1-1 μM olaparib at 1% cell survival (BRAF-mutant: P ≤ 0.05; BRAF-WT: P ≤ 0.001), calculated by one-way ANOVA in multiple comparison tests). Data show the mean of n = 3 experiments ± SEM (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Both Chk1 inhibitors, and trametinib, were also effective radiosensitisers in the majority of cell lines tested.Vemurafenib was ineffective in BRAF WT (IC50frequently not reached), but showed some efficacy in BRAF mutated cell lines, (not significant for radiosensitisation). This limited effect may arise from feedback activation of EGFR, PI3K or alternative signaling pathways, reducing vemurafenib efficacy in CRC when compared to melanoma37.
Validation of radiosensitisation by PARP inhibitors with clonogenic survival assays
As PARPi were the most effective radiosensitisers of the CRC cell line panel, clonogenic survival assays were used to measure radiation enhancement ratios (RERs) in 3 cell lines that were strongly radiosensitised (> 10-fold IC50shift) and 3 cell lines with IC50shift < 10-fold. To potentially improve PARPi radiosensitisation of these resistant cell lines, a more trapping PARPi, talazoparib, was included in these assays.Survival curves (Figure 3), and RERs (Table 3) reflected the proliferation assay results: Olaparib and rucaparib significantly radiosensitised RKO, DLD1, and HT29 compared to vehicle-treated cells, while radiosensitisation of HT55, Colo678, and C10 was limited - although significant for HT55 cells treated with rucaparib. Talazoparib significantly radiosensitised all cell lines tested, and was overall the most effective radiosensitiser (average RERs 1.21-1.92), followed by rucaparib (average RERs 1.15-1.41)and finally olaparib (average RERs 1.12-1.4).
To indicate potential normal tissue toxicity, PARPi experiments were repeated in three non-malignant cell lines,HFLA, MRC5 and RPE. In clonogenic assays (Table 3), these non-malignant cells were significantly radiosensitised by talazoparib. Radiosensitisation by rucaparib was significant for HFLA and MRC5, and radiosensitisation by olaparib was significant only for MRC5 cells (P ≤ 0.05).
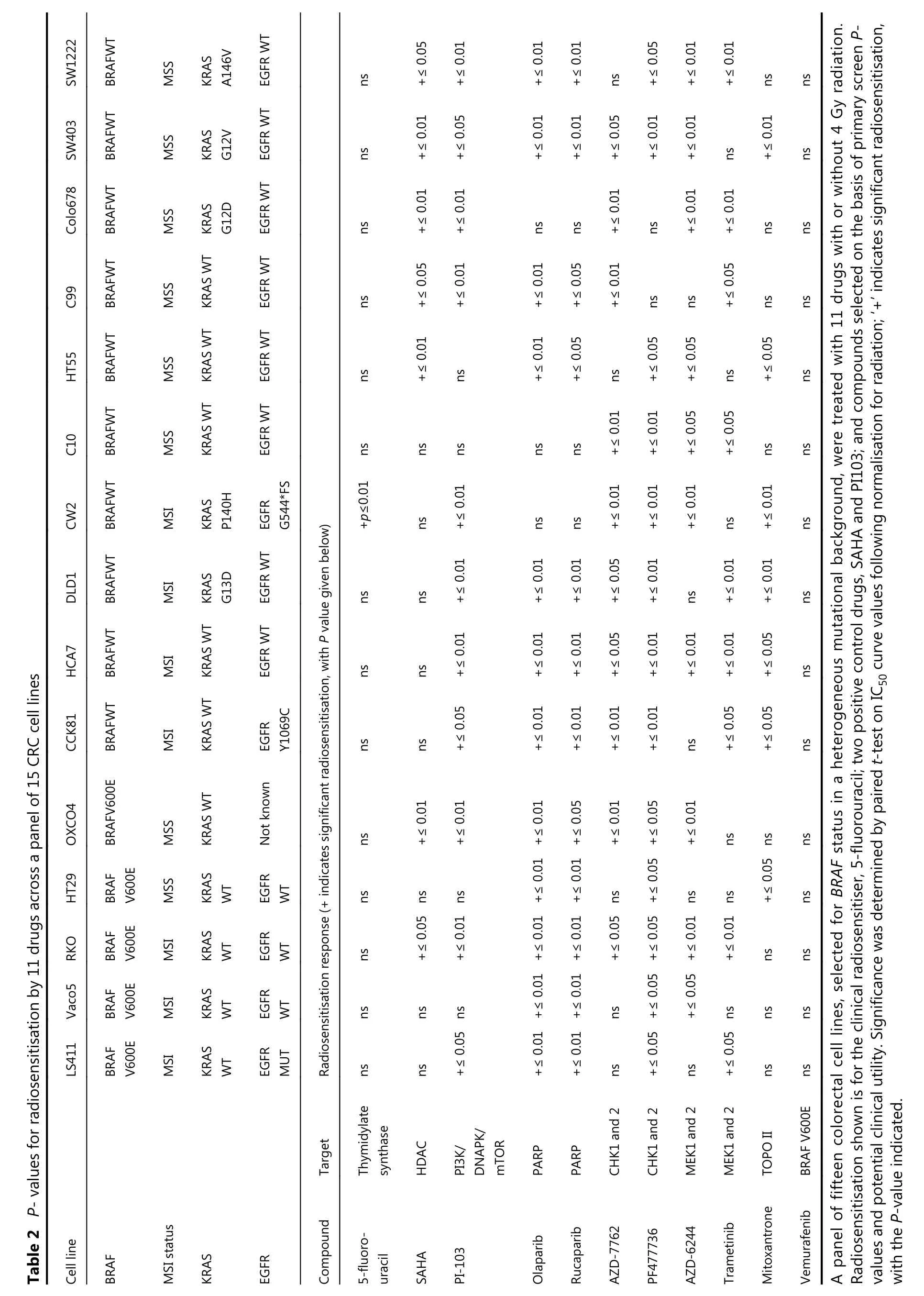
SW1222 BRAFWT MSS KRAS A146V EGFR WT ns +≤ 0.05+≤ 0.01+≤ 0.01+≤ 0.01 ns +≤ 0.05+≤ 0.01+≤ 0.01 ns ns SW403 BRAFWT MSS KRAS G12V EGFR WT ns +≤ 0.01+≤ 0.05+≤ 0.01+≤ 0.01+≤ 0.05+≤ 0.01+≤ 0.01 ns +≤ 0.01 ns Colo678 BRAFWT MSS KRAS G12D EGFR WT ns +≤ 0.01+≤ 0.01 ns ns +≤ 0.01 ns +≤ 0.01+≤ 0.01 ns ns C99 BRAFWT MSS KRAS WT EGFR WT ns +≤ 0.05+≤ 0.01+≤ 0.01+≤ 0.05+≤ 0.01 ns ns +≤ 0.05 ns ns HT55 BRAFWT MSS KRAS WT EGFR WT ns +≤ 0.01 ns +≤ 0.01+≤ 0.05 ns +≤ 0.05+≤ 0.05 ns +≤ 0.05 ns C10 BRAFWT MSS KRAS WT EGFR WT ns ns ns ns ns +≤ 0.01+≤ 0.01+≤ 0.05+≤ 0.05 ns ns CW2 BRAFWT MSI KRAS P140H EGFR G544*FS+p≤0.01 ns +≤ 0.01 ns ns +≤ 0.01+≤ 0.01+≤ 0.01 ns +≤ 0.01 ns DLD1 BRAFWT MSI KRAS G13D EGFR WT ns ns +≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.05+≤ 0.01 ns +≤ 0.01+≤ 0.01 ns Table 2 P- values for radiosensitisation by 11 drugs across a panel of 15 CRC cell lines HCA7 BRAFWT MSI KRAS WT EGFR WT ns ns +≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.05+≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.05 ns CCK81 BRAFWT MSI KRAS WT EGFR Y1069C ns ns +≤ 0.05+≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.01 ns +≤ 0.05+≤ 0.05 ns OXCO4 BRAFV600E MSS KRAS WT Not known ns +≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.01 ns ns HT29 BRAF V600E MSS KRAS WT EGFR WT ns +≤ 0.05ns ns RKO BRAF V600E MSI KRAS WT EGFR WT ns +≤ 0.05ns+≤ 0.01ns+≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.01+≤ 0.05+≤ 0.05ns+≤ 0.05+≤ 0.05+≤ 0.05+≤ 0.01ns+≤ 0.01ns ns ns Vaco5 BRAF V600E MSI KRAS WT EGFR WT ns ns ns +≤ 0.01+≤ 0.01 ns +≤ 0.05+≤ 0.05 ns ns ns LS411 BRAF V600E MSI KRAS WT EGFR MUT Radiosensitisation response (+ indicates significant radiosensitisation, with P value given below)ns ns +≤ 0.05+≤ 0.01+≤ 0.01 ns +≤ 0.05 ns +≤ 0.05 ns ns Thymidylate Target synthase HDAC DNAPK/mTOR PARP PARP CHK1 and 2 CHK1 and 2 MEK1 and 2 MEK1 and 2 TOPO II BRAF V600E Cell line BRAF KRAS MSI status EGFR Compound PI3K/5-fluorouracil SAHA PI-103 Olaparib Rucaparib AZD-7762 PF477736 AZD-6244 Trametinib Mitoxantrone Vemurafenib A panel of fifteen colorectal cell lines, selected for BRAF status in a heterogeneous mutational background, were treated with 11 drugs with or without 4 Gy radiation.Radiosensitisation shown is for the clinical radiosensitiser, 5-fluorouracil; two positive control drugs, SAHA and PI103; and compounds selected on the basis of primary screen Pvalues and potential clinical utility. Significance was determined by paired t-test on IC50 curve values following normalisation for radiation; ‘+' indicates significant radiosensitisation,with the P-value indicated.
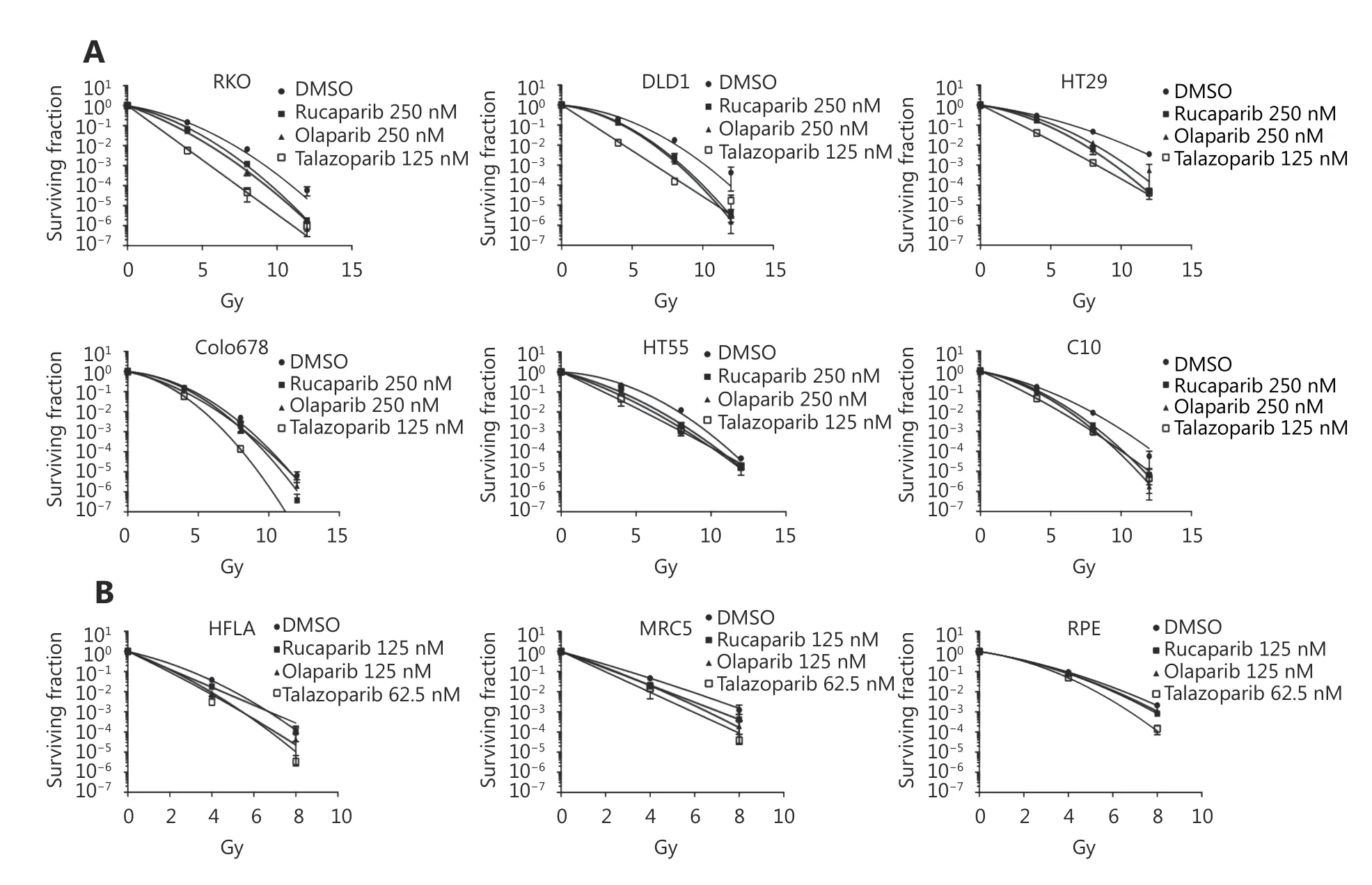
Figure 3 Clonogenic assays to confirm radiosensitisation of multiple cell lines by PARP inhibitors. (A) Colorectal cancer cell lines were plated, rested overnight, drugged and 6 hours later, the cells were either mock irradiated, or irradiated at 4, 8, or 12 Gy. Separation between the control (DMSO) and treated curves indicates radiosensitivity induced by the compound. (B) Human lung fibroblast (HFLA and MRC5)and retinal epithelial (RPE) non-malignant cell lines were drugged in an identical manner and irradiated with 0, 4 or 8 Gy to determine noncancer cell survival following similar treatment. Data show mean of n=3 experiments±SEM.
Validation of PARP inhibitors as radiosensitisers in xenograft studies
The PARP inhibitor talazoparib was the most effective radiosensitiser and had not previously been tested with radiotherapy in animal CRC models. To confirm the in vitro radiosensitisation by PARPi in an in vivo model, talazoparib was tested against two cell lines that were effectively radiosensitised by the drug in 2D assays. Mice were inoculated with subcutaneous tumors consisting of RKO or DLD1 cells, and treated with talazoparib or vehicle, either alone or one hour before each of 2 × 5 Gy radiation treatments. In DLD1 cells (Figure 4A), single treatment with talazoparib or radiation alone did not inhibit tumour growth.Combined talazoparib/radiation treatment was tolerated by the mice, and significantly reduced tumour growth compared with radiation alone (P ≤ 0.01). For the RKO cell xenograft model, there was no significant difference between the effect of radiation alone, and the radiation/talazoparib combination. Tumour histology, levels of perinecrotic hypoxia (CA9 staining) and necrosis were similar for both cell types (Figure 4B).
Discussion
The aim of this study was to identify treatment options to radiosensitise colorectal cancer cells in the context of key mutations that characterise the disease. Biopsies from CRC patients are routinely screened for BRAF, KRAS and PIK3CA mutations, but this information is not currently used in treatment decisions regarding radiotherapy. There is preclinical evidence that single gene alterations in cancer can determine the extent of radiosensitisation exerted by different drugs. Examples include mammalian AMPactivated protein kinase dependence of pancreatic cancer cells to radiosensitisation by metformin38, the role of mismatch repair deficiency in radiosensitisation of CRC cell lines by gemcitabine39-40and p53-dependent radiosensitisation by valproic acid41. Radiosensitisation drug discovery across different genetic backgrounds may enable a change from a “one size fits all” chemo- radiotherapy to the identification of the most appropriate drugs for radiotherapy based on the genetic profile of the cancer.
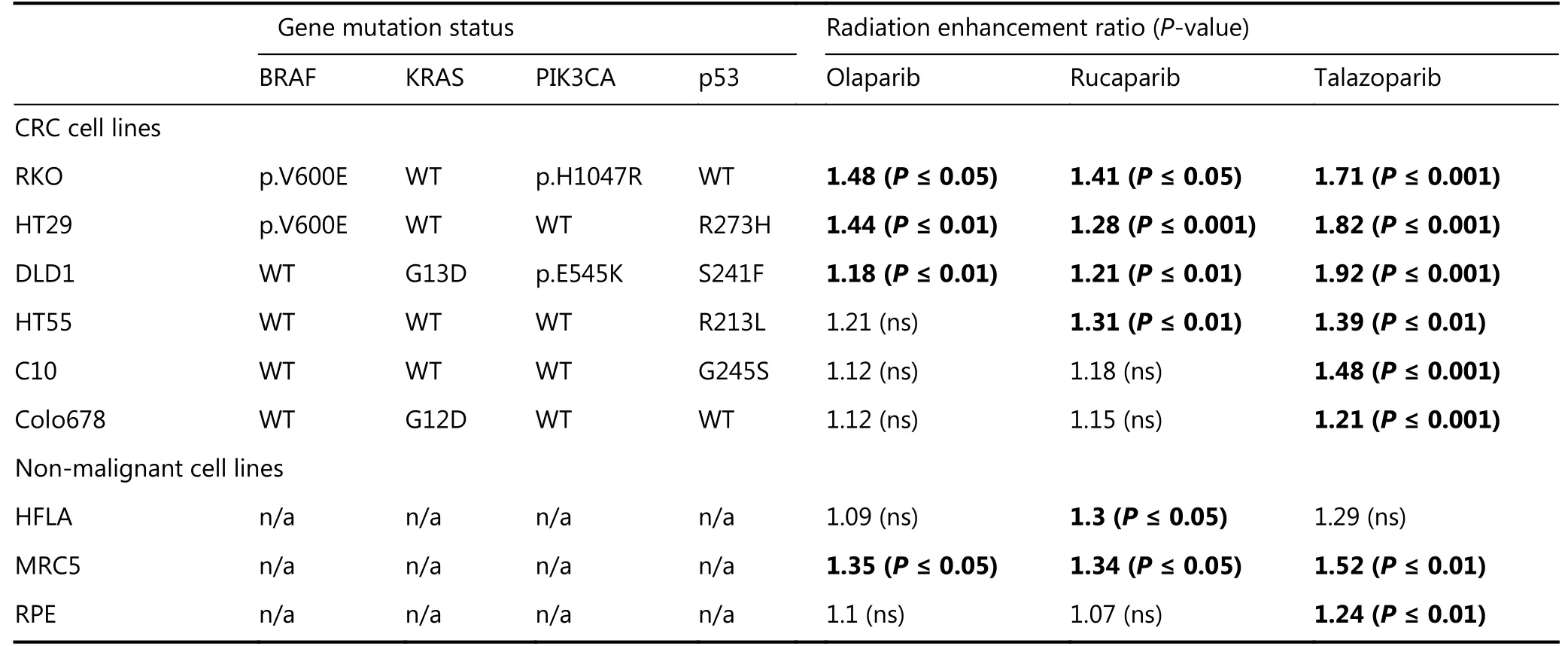
Table 3 Radiation enhancement ratios of PARP inhibitors for colorectal cancer and non-malignant cell lines
To address our primary aim, we developed a novel highthroughput screen to test drug library/radiotherapy combination against cell lines. For drug repurposing, which allows more rapid translation in to the clinic, we used a library of drugs already in clinical use or in clinical trials.Previous investigators using more focused library screens have successfully identified radiosensitisers of CRC42and our study identified the same drugs with radiosensitising potential, the CHK inhibitor, AZD-7762, and the dual mTOR/PI3K inhibitor, dactosilib. We initially used isogenic cell lines to identify radiosensitisers active in a BRAF V600E background. Reassuringly, our results confirmed radiosensitisation by agents from drug classes previously shown to have radiosensitising activity in other published papers, such as inhibitors of the RAS/MEK/ERK, and PI3K/MTOR pathways. In addition, we identified compounds not previously known to be radiosensitisers(Table 1). Of the drugs targeting mutated BRAF(vemurafenib, dabrafenib, RAF265), only vemurafenib reached the threshold for hit-detection in the screen, possibly because vemurafenib is a more potent radiosensitizer, at least compared with dabrafenib43.
Cell lines manipulated by gene mutation might not be entirely representative of the molecular landscape of cancer in patients. We therefore validated results from isogenic cell lines in a panel of human colorectal cancer cell lines,inclusive of common CRC mutations and previously shown to be a useful model for drug development22,44. This approach was also novel since this cell line panel has not previously been used to test new drug-radiotherapy combinations. The results (shown in Table 2), confirmed PARPi as significant radiosensitisers, notably across a much broader range of cell lines than 5FU, the current clinical standard, suggesting that 5FU may not be the optimal treatment for all CRC patients compared to newer and more targeted drugs. This reflects data in other studies in CRC,which show that radiosensitisation by 5FU varies depending on the cell line used45,46. Additionally, the timing of 5FU exposure may influence the degree of radiosensitisation47.
In future, immunotherapy is likely to be of increasing importance in CRC treatment, although at present it is only used to treat the more immunogenic MSI-high tumours48.Despite this, radiotherapy is likely to remain an important treatment for rectal cancer and metastatic disease,particularly when the cost effectiveness of treatment is considered. The broad range of cell lines for which PARPi appear to be suitable radiosensitisers in this study may predict its potential future utility in a wide patient population.
Three PARPi, olaparib, rucaparib, and niraparib, have been approved by the US FDA for the treatment of ovarian cancer, including BRCA-deficient tumours that have deficient homologous recombination repair. PARPi function by inhibiting the binding, or enzymatic activity, of PARP to single strand breaks in DNA. The absence of SSB repair leads to double strand break (DSB) formation at the approaching replication fork, and cell death. It has been shown that PARPi have an increased radiosensitising effect on DSB- repair deficient tumour cells compared with DSB- repair proficient lines49. Compared to olaparib and rucaparib, we found that talazoparib treatment led to higher RERs. PARPi affect cell proliferation by two main actions: inhibiting PARP enzymatic function, and by binding (‘trapping’) PARP to DNA50. Olaparib and rucaparib function primarily through inhibiting enzymatic function, whereas talazoparib ‘traps’PARP at DNA damage sites, with increased anti-proliferative effect, potentially contributing to more effective radiosensitisation51,52.
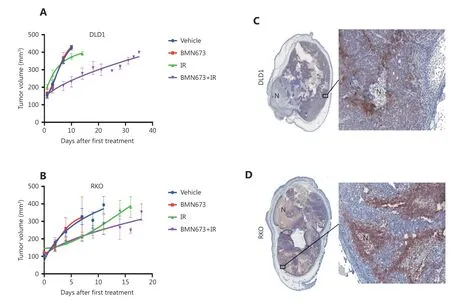
Figure 4 Talazoparib significantly enhances the response of colorectal cancer cells grown in vivo to ionizing radiation. (A, B) Growth of tumour cells injected subcutaneously into the back of BALB/c nude mice, treated as indicated with either; vehicle; 0.1 mg/kg talazoparib;radiation (2 x 5 Gy); or 0.1 mg/kg talazoparib 1 hour prior to each of 2 x 5 Gy radiation doses. Treatment with talazoparib+radiation significantly slowed tumor growth for (A) DLD1 cells but not (B) RKO cells. (C, D) Representative images of tumours harvested from the vehicle treated group (at 400 mm3) show similar histology for both (C) DLD1 and (D) RKO xenografts including perinecrotic hypoxia (CA9 staining, brown) and tumor necrosis (N).
We proceeded to show that the PARP inhibitor,talazoparib, radiosensitised DLD1 xenografts in vivo. The combined treatment caused a prolonged tumour growth delay, in excess of the effects demonstrated elsewhere for combined 5FU/radiation treatment for HCT11645and WiDr53CRC xenografts. It is unclear why talazoparib did not significantly radiosensitise BRAF mutated RKO xenografts in vivo. It has been shown that BRAF-mutant early neoplastic lesions have upregulation of gene sets involved in aberrant DNA methylation54and that BRAF-mutant cancers can have distinct tumour-associated-stroma and components of the extracellular matrix that are different from wild-type cancers55. These complexities may explain the discrepancy between the highly significant results we obtained in 2D culture and the non-significant results we obtained in vivo using the same cell line. Future studies should consider the use of other models, such as patient-derived xenografts or immunocompetent mouse models, to explore this discrepancy further.
Some investigators advocate preclinical comparison of non-malignant with malignant cell lines to identify cancerspecific drugs56,57. In our study, olaparib did not cause significant radiosensitisation of two non-malignant cell lines,HLA and RPE. An in vivo study of intestinal crypt damage, in which fractionated radiotherapy was combined with olaparib, did not appear to cause additional gut toxicity compared to radiotherapy without drug58. Contrastingly,clinical studies of PARPi have documented bowel toxicities as side effects of treatment59and total body irradiation of a p21-reporter mouse has shown that olaparib can exacerbate DNA damage in normal tissues when combined with radiation60. It should be noted that, in our study, rucaparib and talazoparib caused significant radiosensitisation of 2 non-malignant cells tested by clonogenic survival assays. Although talazoparib has already completed phase I development as a single agent61,we recommend that the normal tissue toxicity from the combination of PARPi with radiotherapy should be assessed further in preclinical normal tissue toxicity models and monitored closely in early-phase clinical trials.
In conclusion, our novel approach to radiosensitisation drug discovery in cells isogenic for the BRAF V600E mutation, has led to the identification of PARPi as radiosensitisers for CRC. Validation in a broad panel of human CRC cell lines, and an in vivo xenograft model, has shown potentially broader radiosensitising activity than the current clinical standard of care, 5FU. Following toxicity evaluation of the combination of PARPi with radiotherapy in other preclinical models, we propose that PARP inhibition should be tested in combination with radiotherapy for rectal cancer or metastatic CRC treatment, with careful monitoring of potential toxicities.
Acknowledgements
This work was supported by Bowel Disease Research Foundation, Oxford Cancer Research Centre, the National Institute for Health Research University College London Hospitals Biomedical Research Centre, the Cancer Research UK University College London Experimental Cancer Medicine Centre, CRUK-UCL Centre Award (Grant No. C416/A25145), the Cancer Research UK Centers Network Accelerator Award Grant (Grant No. A21993) to the ART-NET Consortium,and the NIHR Oxford Biomedical Research Centre.
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年2期
Cancer Biology & Medicine2019年2期
- Cancer Biology & Medicine的其它文章
- Effects of palbociclib on oral squamous cell carcinoma and the role of PIK3CA in conferring resistance
- LAG-3 expression on tumor-infiltrating T cells in soft tissue sarcoma correlates with poor survival
- Genetic polymorphisms and gastric cancer risk: a comprehensive review synopsis from meta-analysis and genome-wide association studies
- Hepatitis B virus X protein enhances hepatocarcinogenesis by depressing the targeting of NUSAP1 mRNA by miR-18b
- Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer
- Factors associated with upstaging in patients preoperatively diagnosed with ductal carcinoma in situ by core needle biopsy