
Genetic polymorphisms and gastric cancer risk: a comprehensive review synopsis from meta-analysis and genome-wide association studies
2019-06-18 07:06:14JieTianGuanchuLiuChunjianZuoCaiyangLiuWanlunHeHuanwenChen
Cancer Biology & Medicine 2019年2期
Jie Tian, Guanchu Liu, Chunjian Zuo, Caiyang Liu, Wanlun He, Huanwen Chen
Department of Cardiothoracic Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400010,China
ABSTRACT Objective:In the past few decades, more than 500 reports have been published on the relationship between single nucleotide polymorphisms (SNPs) on candidate genes and gastric cancer (GC) risk. Previous findings have been disputed and are controversial. Therefore, we performed this article to summarize and assess the credibility and strength of genetic polymorphisms on the risk of GC.Methods:We used Web of Science, PubMed, and Medline to identify meta-analyses published before July 30th, 2018 that assessed associations between variants on candidate genes and the risk of GC. Cumulative epidemiological evidence of statistical associations was assessed combining Venice criteria and a false-positive report probability (FPRP) test.Results:Sixty-one variants demonstrated a significant association with GC risk, whereas 29 demonstrated no association. Nine variants on nine genes were rated as presenting strong cumulative epidemiological evidence for a nominally significant association with GC risk, including APE1 (rs1760944), DNMT1 (rs16999593), ERCC5 (rs751402), GSTT1 (null/presence), MDM2(rs2278744), PPARG (rs1801282), TLR4 (rs4986790), IL-17F (rs763780), and CASP8 (rs3834129). Eleven SNPs were rated as moderate, and 33 SNPs were rated as weak. We also used the FPRP test to identify 13 noteworthy SNPs in five genome-wide association studies.Conclusions:Sixty-one variants are significantly associated with GC risk, and 29 variants are not associated with GC risk;however, five variants on five genes presented strong evidence for an association upgraded from moderate. Further study of these variants may be needed in the future. Our study also provides referenced information for the genetic predisposition to GC.
KEYWORDS Gastric cancer; genetic variants; susceptibility; meta-analysis; genome-wide association study
Introduction
Gastric cancer (GC) is a malignant carcinoma of the digestive tract and has become the third highest cause of carcinomaassociated deaths worldwide1. Although advances in diagnoses and treatment may reduce mortality and morbidity, the 5-year survival rate remains poor2. In 2016,26,370 patients had GC, and 10,730 patients died from GC in the United States3. Because the carcinogenic mechanism of GC is not fully understood, GC has affected public health and has become a common concern. As with other complex diseases, the development of GC is a complicated, multistep,multifactorial process, with various potential risk factors,including tobacco use, diet, alcohol consumption,Helicobacter pylori (HP) infection, obesity, and a history of stomach disorder4. In addition, the development of GC may be related to genetic susceptibility factors5-8,12-14. Single nucleotide polymorphisms (SNPs), a common type of genetic mutation, may accelerate the development of GC.Genome-wide association studies (GWAS) may be able to identify sequence variations in the human genome, screen SNPs related to human diseases5, and extend our understanding of associations between genetic variations and cancer risk6. To date, two-stage GWAS (discovery and replication) have discovered millions of SNPs and identified relationships between candidate-gene SNPs and disease susceptibility7,8. There are two limitations associated with the study of candidate genes: small sample size and low statistical power. A more precise and true association can be observed via a meta-analysis using previously available results9,10. In 2008, Dong et al.11performed a meta-analysis and reported that six variants were significantly associated with the risk of GC. Subsequently, more GC-related genes or loci have been discovered in research on genetic variants and identified in GWAS. In 2010, Abnet et al.12identified two identified GCrelated loci (3q13.31 and 5p13.1). In 2011, Jin et al.13found a locus (6p21.1) associated with GC in a GWAS. Later, Hu et al.14identified a new locus (1q22) associated with GC, and Wang et al.15found two novel loci (5q14.3 and 8q24.3)associated with GC. Recently, the results of most metaanalyses for same variant have been inconsistent, suggesting the possibility of false positive associations. Although metaanalyses involving large numbers of patients may reflect true associations between genetic variants and GC risk, the credibility and strength of these associations need to be further assessed16.
Therefore, a comprehensive review associated with genetic susceptibility to GC is needed. Our study assesses the credibility and strength of significant associations between candidate-gene SNPs and GC risk and provides comprehensive information for further investigation.
Methods
Literature search
Web of Science, PubMed, and Medline were searched to identify relevant meta-analyses published on before July 30th,2018 using the following terms: (“gastric”) and (“cancer” or“carcinoma” or “adenocarcinoma” or “tumor” or“malignant” or “malignancy” or “neoplasm” or “neoplasia”or “oncology”) and (“genetic” or “gene” or “polymorphism”or “SNP” or “single nucleotide polymorphism” or “genetic variant”) and (“meta-analysis”). Additionally, we examined all relevant references to identify potential meta-analyses that could offer relevant data.
Criteria for selection
We used the following criteria to screen meta-analyses : (i)publications were in English, (ii) cancer type was GC, (iii)patients with GC were pathologically or histologically confirmed, (iv) the sample size was not fewer than 1,000, and(v) the studies were on GC incidence (rather than mortality or survival rate). Meta-analyses of GWAS were obtained from PubMed. We used the following criteria for GWASrelated articles: (i) publications were in English; (ii) cancer type was GC, which includes all GC subtypes; (iii) patients with GC were pathologically or histologically confirmed; (iv)the studies were on GC incidence (rather than mortality or survival rate); and (v) the studies included two phases(discovery and replication).
Data extraction
Two authors (J.T. and G.L.) worked together to extract all data. Any disagreement was resolved by further discussion.The publication details collected included: first author,publication year, gene name, genetic variant, odds ratio(OR), and 95% confidence intervals (CIs) under the additive model, number of studies, number of subjects (cases and controls), ethnicity of participants, I-square (I2),heterogeneity (Q test)17, and publication bias (Egger’s test)18.For GWAS, the following inclusion criteria were reported for SNPs: (i) the results contained two stages (discovery and replication), (ii) the OR and 95% CI could be collected, and(iii) the P value was less than the cutoff of 5 × 10-8. The publication details collected for each eligible qualified SNP included: PubMed identifier (PMID) number, first author,publication year, gene name, genetic variant, ethnicity of participants, number of subjects (cases and controls), minor allele frequency (MAF), OR and 95% CI, and P-value.
The eligible studies reported two major ethnicities: Asian and Caucasian. Twenty meta-analyses reported a single ethnicity; however, several others reported “diverse populations” indicative of two or more ethnicities. Some studies grouped their results by ethnicity, providing data on subgroup analyses. If the same genetic variant was reported in more than one study, we selected the most recently published study with the greatest number and most integrated participants. Current gene names were used to identify the different variants. A P-value of 0.05 or less was considered significant. Many studies utilized different genetic models; we selected the additive model (Supplementary Table S1) as our unified model to extract data and mitigate selection bias. As we were unable to use certain SNPs (n = 20)in the additive model, dominant, recessive, and homozygous models were also used where necessary.
Evaluation of cumulative evidence
We employed the Venice criteria to assess the epidemiological credibility of significant associations identified by the meta-analyses25. Credibility was rated as strong, moderate, or weak (grade A, B, or C, respectively)according to three factors: amount of evidence, replication of association, and protection from bias. Evidence was evaluated by summing the number of alleles or genotypes among cases and controls and divided into three groups:greater than 1000, 100-1000, and less than 100, representing grades A, B, and C, respectively. Certain test allele numbers or genotype amounts could not be extracted; in these cases,we searched the MAF from the NCBI SNP database (dbSNP)to calculate the amounts. Association replication was calculated using heterogeneity statistics assigning one of three grades: grade A (I2< 25%), grade B (25% < I2< 50%), or grade C (I2> 50%). Bias was evaluated using the P-value for publication bias; grade A indicated no observed publication bias (P > 0.05), grade B indicated bias accompanied by a lack of information for identification of evidence, and grade C indicated that bias was statistically evident (P < 0.05). The magnitude of association was related to protection from bias;a summary OR less than 1.15 was graded as a C for an association, unless several studies had identified that the association was replicated prospectively with an absence of publication bias. Cumulative epidemiological evidence of significant associations was assigned one of three levels:strong (A was assigned to all three grades), weak (C was assigned to any grade), or moderate (all other combinations).
We performed a false positive report probability (FPRP)assay with a prior probability of 0.001 and an FPRP cut-off value of 0.2 to uncover potential false positive results among significant associations, and to evaluate whether these associations should be omitted, as suggested by Wacholder et al.19. Statistical power and FPRP values were calculated by the Excel spreadsheet offered on website (http://jncicancerspectrum.oupjournals.org/jnci/content/vol96/issue6). If the calculated FPRP value was below the prespecified noteworthiness value of 0.2, we would consider the association noteworthy,indicating that the association might be true. FPRP evidence was categorized according to three levels: strong (FPRP <0.05), moderate (0.2 ≤ FPRP ≤ 0.05), or weak (FPRP > 0.2).An FPRP less than 0.05 triggered an upgrading of cumulative evidence from moderate to strong or from weak to moderate.Conversely, an FPRP greater than 0.2 triggered a downgrading of cumulative evidence from strong to moderate or from moderate to weak.
Results
Characteristics of the articles included in our study
Our search yielded 1,041 articles (Figure 1). Of these, 308 articles were excluded as duplicates, 457 articles were excluded as irrelevant (not related to genetic variants or GC)after screening the titles and abstracts; the remaining 276 eligible articles were assessed for full-text review. We further excluded 33 non-meta-analyses, 19 non-genetic polymorphisms, 18 mortality or survival studies, 14 studies without an overall meta-analysis, and 28 studies with insufficient data. Twenty-two studies were screened from the reference publication. Subsequently, 186 meta-analyses were eligible for review; these identified 61 variants associated with the risk of GC. In addition, 18 variants (29.5%) were discovered in 2017.
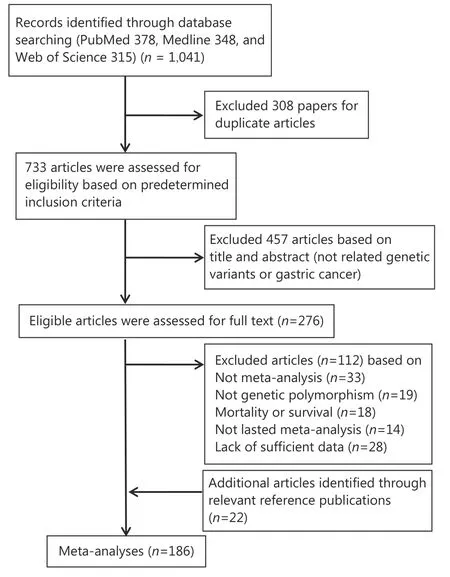
Figure 1 Flow diagram of search strategy and study selection
PubMed was used to identify GWAS related to GC etiology, resulting in a total of five GWAS. All 13 SNPs identified were located within eight genes.
Significant associations in meta-analyses and GWAS
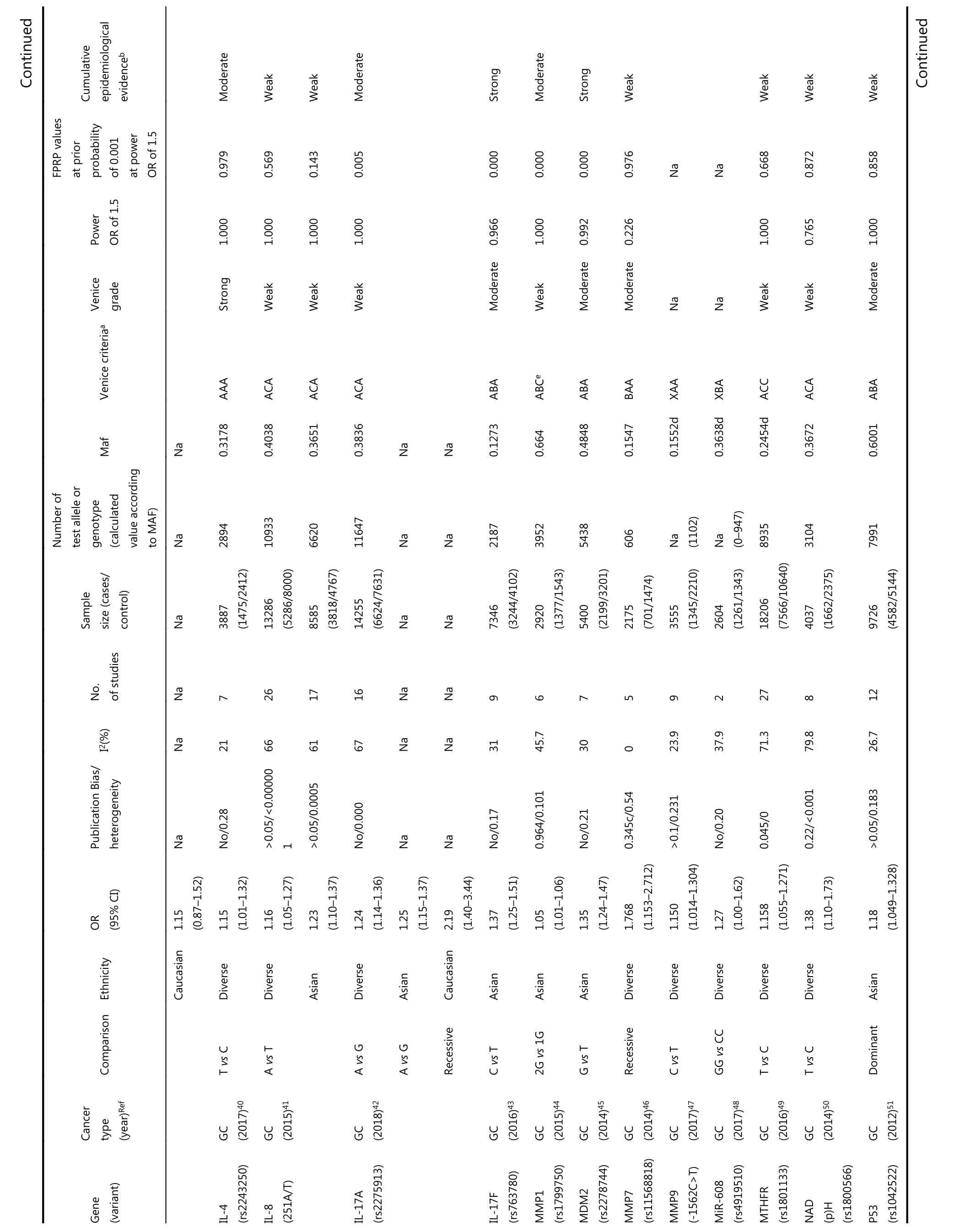
Among the main meta-analyses, cumulative epidemiological evidence was graded for 61 significant associations (Table 1).We assessed these associations using Venice criteria. With regards to the amount of evidence, 41 SNPs were Grade A, 13 Grade B, and 0 Grade C. Based on replication of association,19 were Grade A, 13 were Grade B, and 28 were Grade C.Regarding protection from bias, 52 were Grade A, 7 Grade B,and 0 Grade C. Evidence for association with GC risk was thereby considered strong for seven SNPs, moderate for 17 SNPs, and weak for 29 SNPs based on Venice criteria.
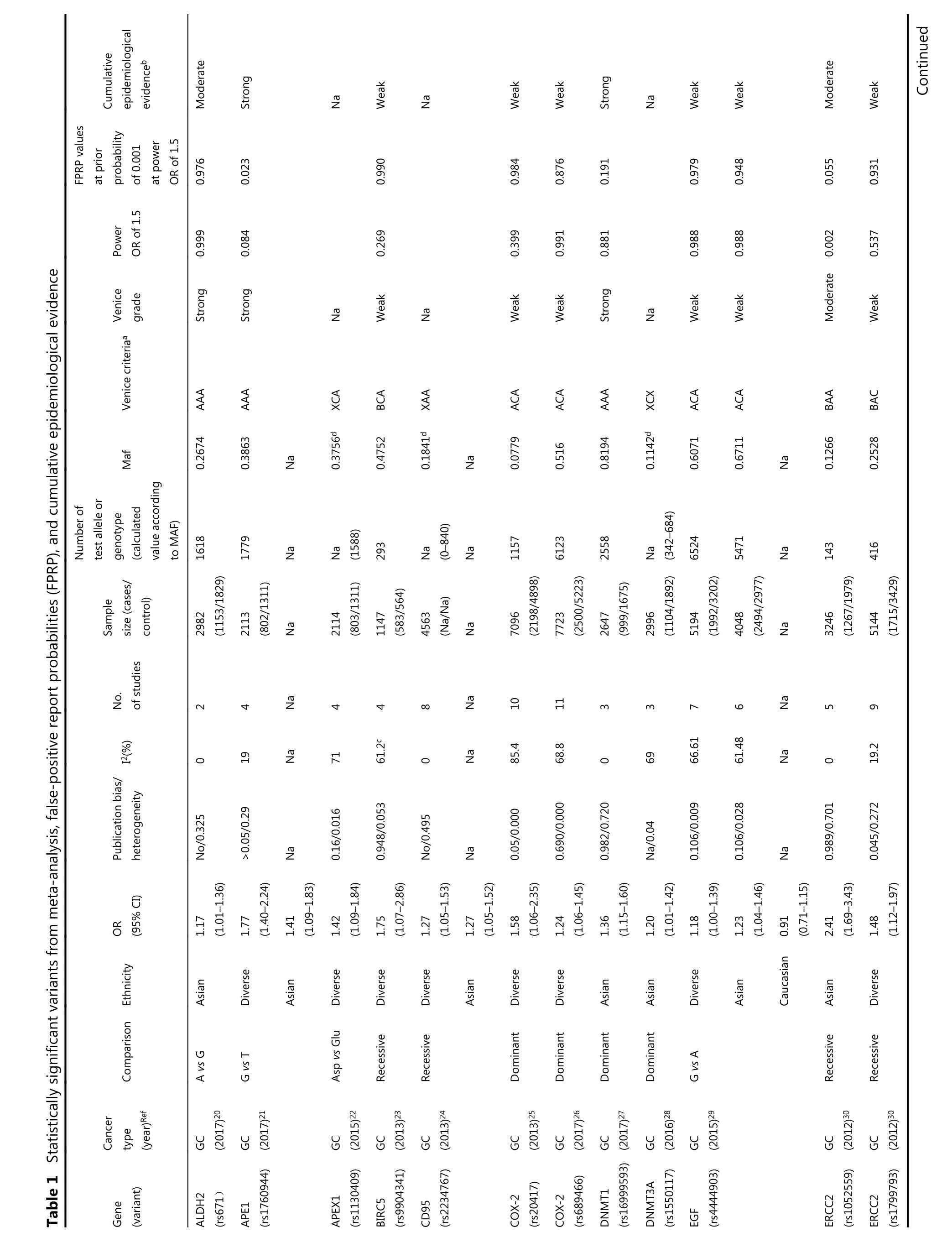
?Continued Table 1 Statistically significant variants from meta-analysis, false-positive report probabilities(FPRP), and cumulative epidemiological evidence

images/BZ_107_215_340_2266_2998.png
images/BZ_108_215_340_2266_2998.png

images/BZ_109_215_340_2266_2998.png
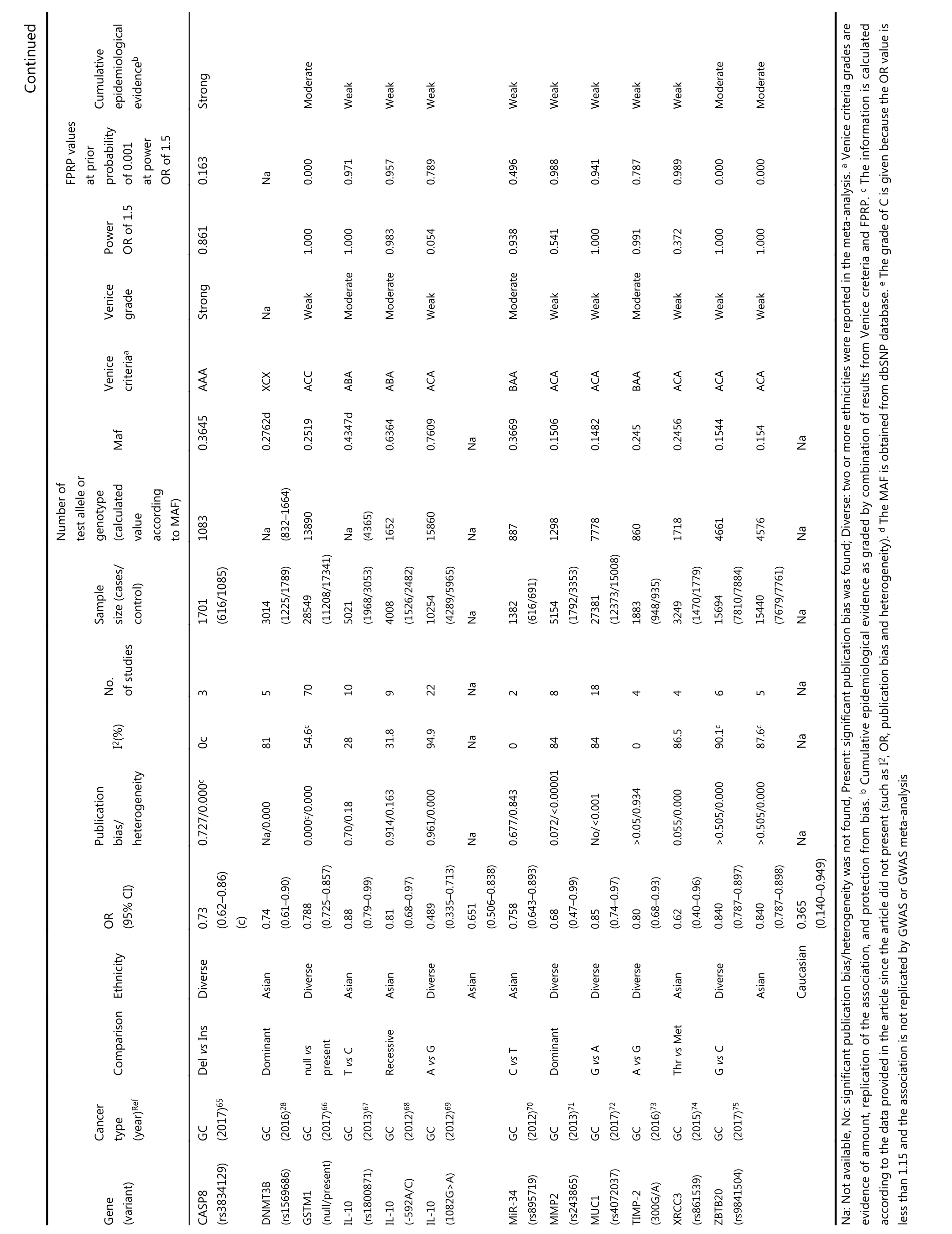
Continued Cumulative epidemiological evidenceb Strong Moderate Weak Weak Weak Weak Weak Weak Weak Weak Moderate Moderate FPRP values at prior probability of 0.001 at power OR of 1.5 0.163 Na 0.000 0.971 0.957 0.789 0.496 0.988 0.941 0.787 0.989 0.000 0.000 Power OR of 1.5 0.861 1.000 1.000 0.983 0.054 0.938 0.541 1.000 0.991 0.372 1.000 1.000 Venice grade Strong Na Weak Moderate Moderate Weak Moderate Weak Weak Moderate Weak Weak Weak Venice criteriaa AAA XCX ACC ABA ABA ACA BAA ACA ACA BAA ACA ACA ACA Maf 0.3645 0.2762d 0.2519 0.4347d 0.6364 0.7609 Na 0.3669 0.1506 0.1482 0.245 0.2456 0.1544 0.154 Na Number of test allele or genotype(calculated value according to MAF)1083 Na(832-1664)13890 Na(4365)1652 15860 Na 887 1298 7778 860 1718 4661 4576 Na Sample size (cases/control)1701(616/1085)3014(1225/1789)28549(11208/17341)5021(1968/3053)4008(1526/2482)10254(4289/5965)Na 1382(616/691)5154(1792/3353)27381(12373/15008)1883(948/935)3249(1470/1779)15694(7810/7884)15440(7679/7761)Na No.of studies 3 5 70 10 9 22 Na 2 8 18 4 4 6 5 Na I2(%)0c 81 54.6c 28 31.8 94.9 Na 0 84 84 0 86.5 90.1c 87.6c Na Publication bias/heterogeneity 0.727/0.000c Na/0.000 0.000c/0.000 0.70/0.18 0.914/0.163 0.961/0.000 Na 0.677/0.843 0.072/<0.00001 No/<0.001>0.05/0.934 0.055/0.000>0.505/0.000>0.505/0.000 Na OR(95% CI)0.73(0.62-0.86)(c)0.74(0.61-0.90)0.788(0.725-0.857)0.88(0.79-0.99)0.81(0.68-0.97)0.489(0.335-0.713)0.651(0.506-0.838)0.758(0.643-0.893)0.68(0.47-0.99)0.85(0.74-0.97)0.80(0.68-0.93)0.62(0.40-0.96)0.840(0.787-0.897)0.840(0.787-0.898)0.365(0.140-0.949)Ethnicity Diverse Asian Diverse Asian Asian Diverse Asian Asian Diverse Diverse Diverse Asian Diverse Asian Caucasian Comparison Del vs Ins Dominant null vs present T vs C Recessive A vs G C vs T Dominant G vs A A vs G Thr vs Met G vs C Cancer type(year)Ref GC(2017)65 GC(2016)28 GC(2017)66 GC(2013)67 GC(2012)68 GC(2012)69 GC(2012)70 GC(2013)71 GC(2017)72 GC(2016)73 GC(2015)74 GC(2017)75 Gene(variant)CASP8(rs3834129)DNMT3B(rs1569686)GSTM1(null/present)IL-10(rs1800871)IL-10(-592A/C)IL-10(1082G>A)MiR-34(rs895719)MMP2(rs243865)MUC1(rs4072037)TIMP-2(300G/A)XRCC3(rs861539)ZBTB20(rs9841504)Na: Not available, No: significant publication bias/heterogeneity was not found, Present: significant publication bias was found; Diverse: two or more ethnicities were reported in the meta-analysis. a Venice criteria grades are evidence of amount, replication of the association, and protection from bias. b Cumulative epidemiological evidence as graded by combination of results from Venice creteria and FPRP. c The information is calculated according to the data provided in the article since the article did not present (such as I2, OR, publication bias and heterogeneity). d The MAF is obtained from dbSNP database. e The grade of C is given because the OR value is less than 1.15 and the association is not replicated by GWAS or GWAS meta-analysis
We then evaluated the probability of a true association with GC risk for the nominally significant variants by calculating their FPRP values. Associations with GC risk showed an FPRP value of less than 0.05 for 12 variants(GSTT1 null/presence, MDM2 rs2278744, PPARG rs1801282,TLR4 rs4986790, IL-17F rs763780, HOTAIR rs920778, IL-17A rs2275913, PRKAA1 rs13361707, PSCA rs2294008,MMP1 rs1799750, ZBTB20 rs9841504, and GSTM1 null/present), 0.05-0.2 for 27 variants, and greater than 0.2 for the remaining 14 variants. Finally, nine variants on nine genes were rated as demonstrating strong cumulative epidemiological evidence of association with GC risk after combining Venice criteria and FPRP results, including APE1(rs1760944), DNMT1 (rs16999593), ERCC5 (rs751402),GSTT1 (null/presence), MDM2 (rs2278744), PPARG(rs1801282), TLR4 (rs4986790), IL-17F (rs763780), and CASP8 (rs3834129). A moderate association with risk was found for 11 variants, and weak association for 33 variants.Eight variants in our study could not be graded because of significant differences between calculated and true amounts.Calculated amounts of less than 3,000 were not included for assessment in MAFs obtained from the dbSNP.
In five GWAS, 13 variants were significantly associated with GC risk (Table 2)12-15,76. Eight variants were significantly associated with an increased risk of GC. The opposite associations were found in five variants, all of which were regarded as significant following the FPRP assay. Venice criteria were not applicable to GWAS.
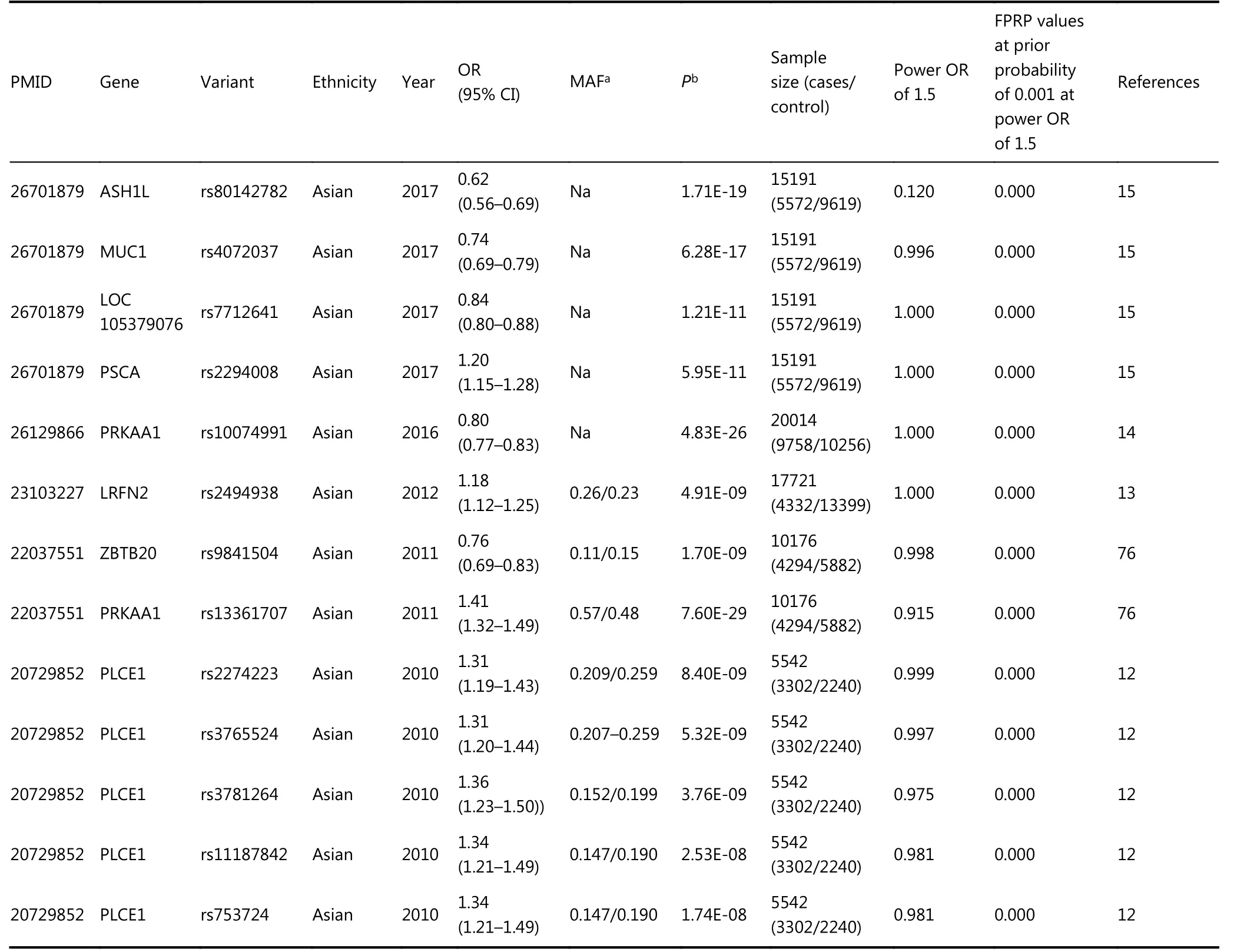
Table 2 Statistically significant variants from GWAS
Non-significant associations in meta-analyses
We performed power analyses to determine the stability of associations. Based on our meta-analysis results, 29 variants were not significantly associated with GC risk24,25,27,32,33,58,63,77-93. Four variants with sample sizes greater than 10,000 were not significantly associated with GC; further investigations into these variants may not be necessary,including hOGG1 Ser326Cys85, IL-1B rs114362786, miR-146a rs291016490, and TGF-β1 rs180046958. Certain variants presented relatively small sample sizes; as such, the evidence for non-association (Supplementary Table S2) was considered unstable.
Inconsistency among meta-analyses
Controversial results were obtained for 23 variants(Supplementary Table S3). Overall, 16 variants were found to be significantly associated with GC, as follows: BIRC5 rs9904341, EGF rs4444903, ERCC5 rs2296147, IL-4 rs2243250, IL-8 251T/A, IL-17F rs763780, MMP7 rs11568818, MMP1 rs1799750, MMP9 -1562C > T, TGF-β1 rs1800470, TNF-α rs361525, DNMT3B rs1569686, GSTM1 null/active, IL-10 -592C > A, IL-10 1082G > A, and MMP2 rs243865. Seven variants were found to be non-significant:CDH1 rs16260, CDH1 +54T > C, hOGG1 Ser726Cys, IL-1B rs1143627, miR-146a rs2910164, miR-196a2 rs11614913, and TGF-β1 rs1800469.
Discussion
In this study, we collated epidemiological evidence demonstrating significant associations between genetic variants and GC risk. We extracted related useful information from meta-analyses and GWAS to support a comprehensive assessment for further evaluation. Using FPRP tests and Venice criteria, we evaluated the credibility of this cumulative epidemiological evidence of nominally significant associations. Nine variants on nine genes were rated as demonstrating strong evidence of association with GC risk, including APE1 rs1760944, DNMT1 rs16999593,ERCC5 rs751402, GSTT1 null/presence, MDM2 rs2278744,PPARG rs1801282, TLR4 rs4986790, IL-17F rs763780, and CASP8 rs3834129. Eleven variants presented moderate evidence of association with GC risk, and 33 variants presented weak evidence.
Apurinic/apyrimidinic endonuclease 1 (APE1), located on chromosome 14q11.2, participates in DNA base excision repair and has been associated with human carcinogenesis94-96. Our analysis provides strong evidence for an association between the G allele of the APE1 polymorphism and GC risk via an additive model, with a 1.77-fold increased risk of GC in a diverse population with a total sample size of 2,113. This variant promotes the development of cancer by impeding DNA repair activity97. In our subgroup analysis,the mutant G allele also increased the risk of GC compared with the wild-type T allele. However, this study was performed exclusively on the Asian population. One reason that concentrated on single (Asian) population could be the small sample size of this meta-analysis, which made subgroup analyses challenging. Therefore, further investigations into this variant are necessary.
DNMT1, located on human chromosome 19p13.2,encodes a protein comprising 1,632 amino acids, which may be associated with the development of carcinoma98. Some studies have suggested that DNA methylation contributes to the progression of GC and that over-expression of DNMT1 may be associated with GC risk. The AKT-NFκB and STAT3 signaling pathways have been implicated in the overexpression of DNMT1, which may cause aberrant DNA methylation on tumor suppressor genes, thereby promoting the progression of GC99,100. There was strong evidence for an association between SNP rs16999593 and GC risk in a sample of 2,647 Asians; this polymorphism occurs on the C allele of DNMT1 (OR = 1.36, 95% CI = 1.15-1.60). This variant results in a histidine to arginine substitution at position 97(His97Arg) of the translated sequence, which may disrupt the function of DNMT1, thus increasing susceptibility to GC.This study sample was limited to Asian populations, and involved a large proportion of Chinese patients. Further studies should investigate this polymorphism in other ethnic groups.
ERCC5, also known as XPG, is an endonuclease that may prevent carcinogenesis by excising damaged DNA during the DNA repair process101. A polymorphism (rs751402) is found in the promoter region of ERCC5 and controls its expression and function during transcription in healthy human cells102.The present study showed that this SNP in a dominant model was strongly associated with increased risk of GC; rs751402,which contains a C to T transition, may alter the transcription domain-associated repair capacity of ERCC5 that could account for its correlation with GC cancer risk in Asians (n = 9,814). However, all studies were performed on a single ethnic group (Asian), and we recommend expanding studies on this polymorphism to other populations.
Human CASP8, located on chromosome 2q33-q34,participates in cell cycle regulation103,104. This SNP(rs3834129), located in the promoter region of CASP8105,leads to reduced expression of this gene. Impaired CASP8 expression can decrease T lymphocyte-induced cell death105.In the additive model, this SNP was strongly associated with GC, with a 1.14-fold decreased risk of GC in the sample population (n = 1,701). The variant inhibits CASP8 transcription by inactivating the binding site of transcription factor stimulatory protein 1105, potentially altering immune surveillance and decreasing the risk of GC. Although this SNP was only evaluated in case-control studies (not in GWAS), its association with GC risk in the meta-analysis was well established, with an overall schema of AAA. We assigned this SNP a rating of strong evidence because of an FPRP value of less than 0.05. This polymorphism could present a novel target for gene therapy of GC and lead to new drug developments against GC.
Five variants were upgraded from moderate to strong because of an FPRP value less than 0.05, including GSTT1 null/presence, MDM2 rs2278744, PPARG rs1801282, TLR4 rs4986790, and IL-17F rs763780. Homozygous deletion (null genotype) of GSTT1 (null/presence) leads to GST enzymatic inactivation and was associated with GC progression in a population of over 20,00035,106,107. Based on these inconsistent results, we assigned strong evidence for the association of this SNP with GC risk, even though this SNP was not evaluated by GWAS. Total samples for the four remaining SNPs were less than 8,000, with 5,400 for MDM2 rs2278744, 1,418 (546/872) for PPARG rs1801282, 5,321 for TLR4 rs4986790, and 7,346 for IL-17F rs763780. According to the results of the FPRP and Venice criteria evaluations,evidence for an association with GC for these four SNPs was not statistically convincing. These results are potentially due to the use of Venice criteria, which accounts for potential bias such as genotyping errors, phenotype misclassifications, and population stratification. Some of the SNPs were difficult to assess using a meta-analysis. It is possible that the results would be more convincing if different weights were assigned to the different categories included in the Venice criteria.
Three SNPs, ALDH2 rs671, FASL rs763110, and IL-4 rs2243250, were rated as being moderately associated with GC risk; all were downgraded from strong to moderate based on an FPRP greater than 0.2. The FPRP method considers the P value, prior probability, and statistical power of the test; as we calculated FPRP at a prior probability of 0.001 and used the statistical power to detect an odds ratio of 1.5 for alleles with an elevated risk in FPRP calculations, some otherwise significant associations may have been excluded. Previous studies using different prior probabilities have classified their results as more noteworthy. Further investigations on these three variants may be necessary to analyze their associations in greater depth.
Seven variants (HOTAIR rs920778, IL-17A rs2275913,PRKAA1 rs13361707, PSCA rs2294008, MMP1 rs1799750,ZBTB20 rs9841504, and GSTM1 null/present) were rated as being moderately associated with GC risk, after being upgraded from weak to moderate based on their FPRP values(< 0.05). Among these variants, rs13361707 on PRKAA1,rs2294008 on PSCA, and rs9841504 on ZBTB20 were evaluated by meta-analysis and GWAS. A high degree of heterogeneity may explain how all three variants were graded“ACA” overall; these variants were designated as having moderate associations with GC. Two SNPs (rs13361707 and rs2294008) increased GC risk by 1.34- and 1.26-fold in the overall study population, respectively. No statistical data were presented for ethnicity subgroups; which could explain the heterogeneity in the data. We recommend subdividing populations by ethnicity to identify potential differences in the association between these two variants and GC. The variant rs9841504 variant was associated with a 1.26-fold decreased risk of GC in the overall population based on a total sample size of 15,694; this sample included a large number of Asian individuals but relatively few individuals of other ethnicities. We found stronger evidence to support an association for this variant in the Asian population based on the large sample size, but not in the smaller mixed-ethnicity group. While ethnicity may be one factor affecting heterogeneity, other factors such as methodology, GC subtypes, and environmental factors may also as account for variation in the data. Further investigations of this and two further variants (MMP1 rs1799750 and HOTAIR rs920778)are necessary, due to a lack of power in the smaller sample sizes.
Twenty-nine variants were not significantly associated with GC risk, including eight variants on five genes and two miRNAs in a sample of approximately 4,000 patients, at approximately 95% power to detect an OR of 1.15 in an additive model for a variant with MAF of 20%. Most of the MAFs of those eight variants exceeded 0.2, despite sample sizes greater than 4,000. We can therefore conclude, that these eight variants are unlikely to be associated with GC(Supplementary Table S4). It is probable that further investigations evaluating these eight variants will not yield meaningful results with regards to GC.
Of the remaining variants, 23 presented inconsistent associations with GC risk, 16 variants were nominally associated with GC risk, and seven variants were conclusively not associated with GC risk. Many studies analyzing the same SNP from this group yielded inconsistent results, due to variation in sample size, selection of association models, and ethnicity. If the same genetic variant was reported in more than one article and the results were not consistent, we selected the most recently published meta-analysis to obtain the highest number or most integrated participants. When selecting association models, the additive one was the model of choice; others were employed only when the additive model was unusable. We extracted information from subgroup analyses based on ethnicity and found that some results for the same variant differed by ethnicity, which may have contributed to inconsistency in the results(Supplementary Table S5). Of note, we found that all GWAS and most of the meta-analyses included in our review were performed in Asian populations. Approximately 40% of all patients with GC worldwide are Asians, with a high proportion found in China. Studies in western populations performed with smaller sample sizes may exist but were not included in our study because of low statistical power.Additional studies on other ethnicities with larger sample groups are strongly recommended for the future.
Certain limitations apply to this report. Although we performed a comprehensive literature search, it is possible that some articles may have been missed. Variability in sample size was found among different studies; smaller sizes may have affected the credibility of the data. We evaluated data extracted from a single source, which may have introduced a critical bias. Finally, we only evaluated the susceptibility to, and incidences of association between genetic variants and GC risk; the involvement of genetic polymorphisms as they contribute to tumor progression,metastasis, and drug resistance in GC were not assessed due to a lack of data or information. Despite these limitations, we believe that our study, which provides an updated summary and evaluation of existing literature on the genetic predisposition to GC, will be of value in informing future genetic studies.
This paper evaluated the cumulative epidemiological evidence of significant associations between genetic variants and GC risk by combining Venice criteria and a FPRP assay.Nine SNPs presented strong evidence for an association with GC, of which five variants on five genes were upgraded from moderate to strong evidence based on their FPRP values, and should be further assessed in future studies. If these nine variants are confirmed to be associated with GC risk, they may explain the partial effect of the genetic variant on GC risk. In summary, our study summarizes current literature on the genetic architecture of GC susceptibility, and provides useful data for designing future studies aiming to assess genetic factors for GC risk.
Acknowledgements
Part of the suggestion for the discussion on statistics and proofreading of English grammar were provided by Dr.Shengping Hou (The First Affiliated Hospital of Chongqing Medical University, Chongqing Eye Institute, Chongqing Key Laboratory of Ophthalmology, Chongqing, China).
Conflicts of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年2期
Cancer Biology & Medicine2019年2期
- Cancer Biology & Medicine的其它文章
- Factors associated with upstaging in patients preoperatively diagnosed with ductal carcinoma in situ by core needle biopsy
- Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer
- Dorsomorphin induces cancer cell apoptosis and sensitizes cancer cells to HSP90 and proteasome inhibitors by reducing nuclear heat shock factor 1 levels
- Effects of palbociclib on oral squamous cell carcinoma and the role of PIK3CA in conferring resistance
- Hepatitis B virus X protein enhances hepatocarcinogenesis by depressing the targeting of NUSAP1 mRNA by miR-18b
- Identification of anticancer drugs to radiosensitise BRAFwild-type and mutant colorectal cancer