
Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer
2019-06-18 07:06:32HuizhiSunNanYaoSiqiChengLinqiLiShiqiLiuZhaoYangGuanjieShangDanfangZhangZhiYao
Cancer Biology & Medicine 2019年2期
Huizhi Sun, Nan Yao, Siqi Cheng, Linqi Li, Shiqi Liu, Zhao Yang, Guanjie Shang, Danfang Zhang,2,Zhi Yao,3
1Department of Pathology, Tianjin Medical University, Tianjin 300070, China;
2Department of Pathology, General Hospital of Tianjin Medical University, Tianjin 300070, China;
3Department of Immunology, Tianjin Medical University, Tianjin 300070,China
ABSTRACT Objective: Vasculogenic mimicry (VM) channels that are lined by tumor cells are a functional blood supply in malignant tumors.However, the role of VM-initiating cells remains poorly understood. Cancer stem-like cells (CSCs) are positively correlated with VM. In this study, triple-negative breast cancer (TNBC) enriched with CSCs was used to investigate the relationship between VM and CSCs.Methods: The expression of several CSC markers was detected by immunohistochemistry in 100 human breast cancer samples.The clinical significance of CSC markers and the relationship between VM, CSCs, breast cancer subtypes, and VM-associated proteins were analyzed. CD133+ and ALDH+ human and mouse TNBC cells were isolated by FACS to examine the ability of VM formation and the spatial relationship between VM and CSCs.Results: CSCs were associated with TNBC subtype and VM in human invasive breast cancer. CSCs in TNBC MDA-MB-231 cells formed more VM channels and expressed more molecules promoting VM than the non-TNBC MCF-7 cells in vitro. MDA-MB-231 cells that encircled VM channels on Matrigel expressed CD133. Moreover, CSCs were located near VM channels in the 3D reconstructed blood supply system in human TNBC grafts. The CD133+ and ALDH+ cells isolated from TA2 mouse breast cancer formed more VM channels in vivo.Conclusions: CSCs line VM channels directly. Additionally, CSCs provide more VM-related molecules to synergize VM formation. The signaling pathways that control CSC differentiation may also be potential treatment targets for TNBC.
KEYWORDS Vasculogenic mimicry; triple-negative breast cancer; cancer stem-like cells; ALDH1; CD133
Introduction
Tumor angiogenesis is an important condition for the growth and evolution of tumors1. Tumor angiogenesis involves host blood vessel proliferation and the formation of new blood vessels1. Angiogenesis in the bone marrow involves endothelial progenitor cells that are recruited into tumors for colonization and differentiation to form vasculature2. Vasculogenic mimicry (VM) involves tumor cells that mimic endothelial cells through deformation and extracellular matrix (ECM) interaction, which surround the lumen and create endothelium-dependent vascular connectivity, becoming functional tumor blood vessels2-4.
At present, VM in melanoma, hepatocellular carcinoma,ovarian carcinoma, glioma, lung cancer, and more than 10 types of malignant tumors has been identified5-9. The presence of VM tumors was associated with a higher degree of malignancy, concerning cell atypia, mitotic figures,invasion, metastasis, and prognosis1. The presence of VM tumors increases the risk of hematogenous metastasis and poor survival prognosis1. In addition, the structure of VM is composed of tumor cells. Traditional anti-angiogenic drugs targeting endothelial cells are not effective in VM and even promote tumor invasion and metastasis10-12. Therefore,finding an effective therapeutic target is the key to improving the prognosis of patients with VM tumors.
The formation of VM is the focus of VM research, such as the ability of VM tumor cells to adopt the pluripotent phenotype of embryonic cells13,14. Cancer stem-like cell(CSC) marker expression associated with VM3in human malignant tumors suggest that CSCs may participate in the formation of VM. However, there is no direct evidence that tumor stem cells are directly involved in the regulation of VM formation.
Previously, VM has been found in human breast cancers,and VM is associated with different breast cancer molecular types10,15. There is increased VM in triple-negative breast cancer (TNBC) compared to non-TNBC10. The molecular characteristics of TNBC are enriched with cancer stem-like cells16,17. In this study, immunohistochemical staining (IHC),multiple fluorescent staining, confocal laser microscopy, and 3D image reconstruction were performed to observe the spatial location relationship between tumor stem cells and VM in TNBC.
Materials and methods
Cells and agents
Human breast cancer MCF-7 cells and MDA-MB-231 cells were purchased from the national experimental cell resource sharing service platform (Beijing). TA2 mouse breast cancer cells were provided by the Animal Department of Tianjin Medical University. The primary antibodies used in this study are listed in Supplementary Table S1. All secondary antibodies were purchased from Zhongshan Golden Bridge Biotechnology Co., Ltd. MCF-7 cells, MDA-MB-231 cells,and TA2 mouse breast cancer cells were cultured in RPMI-1640 medium with 10% FBS, 4 mM L-glutamine, and 1%penicillin-streptomycin. Matrigel (BD Bioscience) was diluted with RPMI-1640 medium in 3D cell culture.
Patient samples
All human studies were approved by the Tianjin General Hospital Ethics Committee (Approval No. TMULA-201821).All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. The participating patients were informed about the aims,methods, and other details of the medical research. Samples were collected from 100 breast cancer patients with detailed pathological and clinical information. All patients underwent surgery and chemotherapy in Tianjin General Hospital from 1997 to 2004. The median age of the patients was 48.0 years(27 to 74 years). All patients had invasive breast cancer, and 45 cases with axillary node metastasis were reported. The follow-up period ranged from the time of the surgery to December 2008.
IHC staining and counting method
IHC was performed on the tissue microarray sections by following a standard protocol. Protein expression was quantified according to the method of Sun et al. For stain intensity, “0” denoted no staining, “1” denoted weakly positive staining, “2” denoted moderate staining, and “3”denoted strong staining. The number of positive cells out of the 100 tumor cells per field was visually evaluated and scored as follows: “0” for < 10% positive cells, “1” for < 25%,“2” for < 50%, and “3” for > 50%. The staining index, or the sum of the stain intensity and the positive cell score, was used to determine the final result for each sample. A sample was defined as positive when the staining index was greater than 1.
Immunohistochemical staining for CD31 and periodic acid schiff (PAS) histochemical double staining
After IHC for CD31, the sections were exposed to 1% sodium periodate for 10 min. They were incubated for 15 min at PAS at 37°C after washing with distilled water for 5 min. After, the sections were counterstained with hematoxylin and observed under a microscope (80i, Nikon).
FACS analysis
The suspended MDA-MB-231, MCF-7, and TA2 cells were fixed in 75% cold ethanol, and 106 cells were incubated with CD133-PE antibody (1:11 dilution, Miltenyi Biotec) solution or isotype control on ice for 15 min before they were washed and resuspended in staining buffer (2% fetal calf serum in PBS). ALDH1+ cells were detected and isolated with the ALDEFLUORTM stem cell isolation kit (STEMCELL Technologies). 1 mL of the suspended tumor cells was added into the test tubes, and 5 μL ALDEFLUORTM DEAB was added into the control tubes. Activated ALDEFLUOR at a 5 μL volume was added into the test tubes, and half of the mixture in the test tubes was then removed into the control tubes. All tubes were incubated at 37°C for 30 min.Approximately 0.5 mL ALDEFLUOR assay buffer was added to resuspend the cells. All cells were analyzed and isolated using FACSAria III (BD Biosciences). Gates were set with isotype controls such that, for each cell, less than 1% of the total cell population was false-positive. The labeled cells were then analyzed (10,000 events), and the data were saved as FACS.
Serum-free suspension culture of stem cells
MDA-MB-231 cells in different subtypes were collected to culture in a serum-free suspension culture system. Poly2-hydroxyethyl methacrylate (10 mg/mL in 100% ethanol) was added into 6-well plates (600 μL/plate). The cells were incubated in 6-well plates coated with polyHEMA in serumfree DMEM/F12 media (0.02 g/mL EGF, 0.02 g/mL bFGF,2%/mL B27). Sphere formation was observed using an inverted microscope (Ts2, Nikon).
Western blot
CD133, ALDH1, E-cadherin, and VE-cadherin protein expression was assessed by western blot. Briefly, total protein was obtained using a lysis buffer (1% SDS, 10 mM Tris-HCl,pH 7.6, 20 μg/mL aprotinin, 20 μg/mL leupeptin and 1 mM AEBSF). Protein concentration was measured using the Bradford method. Approximately 20 μg of protein was separated on an 8% SDS-PAGE gel and blotted onto a PVDF membrane. After blocking with 5% fat-free milk in TBSTween overnight, the membrane was incubated with primary antibodies overnight at 4 °C. After washing with TBS-Tween three times, the membrane was labeled with horseradish peroxidase-conjugated anti-goat or IgG (1:1000) for 1 h at room temperature. The blots were developed with a DAB kit,β-actin was used as an internal control, and the bands for the samples were analyzed using a gel imaging system (Kodak).
Immunofluorescent staining
MDA-MB-231 and MCF-7 cells on Matrigel-coated slides were washed with PBS twice, permeabilized, and fixed in 2%PFA and 0.1% Triton X100 in PBS buffer at 4 °C for 30 min.The slides were then washed three times with PBS and incubated with 10% goat serum in PBS. The cells were then incubated with the primary antibodies at 4 °C overnight,washed three times with PBS for 15 min, and incubated with the secondary antibodies for 2 h at room temperature. The slides were washed with PBS and mounted. All matched samples were photographed using a confocal laser scanning microscope (A1, Nikon).
Animal model
The protocols for the animal experiments were approved by the Tianjin Medical University Ethics Committee. All steps were carefully administered to protect the welfare of the animals and prevent their suffering. Nude mice were purchased from Beijing HFK Bioscience Company.Approximately 5 × 106MDA-MB-231 and MCF-7 tumor cells were subcutaneously injected into the back of six-weekold female mice (n = 10/group). Tumors were measured every 2 d using a standard formula (length × width 2 × 0.52).The nude mice were sacrificed when the MDA-MB-231 and MCF-7 tumor size was close to 0.5 cm3. Fluorescein(494/521)-labeled 2,000,000 MW dextran (D-7137,Molecular Probes™) was injected i.v. 60 min before the mice were sacrificed. Tumors were harvested and fixed in 4%paraformaldehyde for 48 h. The Tientsin Albino 2 (TA2)mice were provided by Animal Center of Tianjin Medical University. Approximately 4 × 105ALDH1+ and CD133+TA2 breast cancer cells were subcutaneously injected into the groin of six-week-old female TA2 mice (n = 10/group,respectively). The TA2 breast cancer-bearing mice were sacrificed when the tumor size reached up to 1 cm3. Tumors were harvested and fixed in 4% formalin for 24 h. Tumors were embedded in paraffin, and 5 μm-thick sections were prepared.
Whole mount staining
Whole mount staining was performed as described. Briefly,fixed tumors were cut into small pieces (100 - 200 μm),digested with proteinase K (20 μg/mL) for 5 min, and subsequently treated with 100% methanol for 30 min at room temperature. Nonspecific binding sites were blocked overnight at 4°C with a blocking buffer (3% skim milk in PBS containing 0.3% Triton X-100, PBST). Tissue sections were incubated overnight at 4°C with a rat endomucin antibody(1:100 dilution in blocking buffer; 11-5851-80, eBioscience),a rabbit CD133 antibody (1:50 dilution in blocking buffer,Biorbyt), and a rabbit ALDH1 antibody (1:100 dilution in blocking buffer, LSBio). Sections were rigorously washed with PBST four times. Tumor tissues were further blocked using the blocking buffer for an additional 2 h before incubation with the secondary antibody. An Alexa Fluor 680-labeled goat anti-rabbit secondary antibody (1:200,Invitrogen) and a Texas red-labeled goat anti-rat secondary antibody (1:200, Invitrogen) were incubated with tissues at room temperature for 2 h, followed by washing with PBST twice. Stained tissue sections were mounted with a Vectashield mounting medium (ZLI.9557, Zhongshan) and were analyzed by confocal microscopy (Nikon A1 Confocal microscope, Nikon). Positive signal density was quantified using four to six random fields at 10× or 20×, from four to five tumors per group.
Statistics
SPSS version 11.0 (Chicago, Illinois, USA) was used to evaluate the data in this study. The χ2test was performed to assess the pathological and clinical characteristics of the TNBC and non-TNBC groups. The survival of the two groups was analyzed through a Kaplan-Meier analysis. The relationships between CSC markers, TNBC-type, VM, and VM-related proteins were analyzed by correlation analysis.The two-tailed Student’s t test was performed to compare the difference of the two groups in the CSC population, protein expression, VM-like channel counting, fluorescence intensity,and tumor weight. The significance level was set at P < 0.05.
Results
Clinical significance of VM and cancer stemlike cells in human breast cancer
In the immunohistochemistry for ER, PR, and HER2 expression, a sample was defined as positive when the staining index was over 1. Among the 100 breast cancer cases in this study, 27 were classified as TNBC and the remaining were non-TNBC. Figure 1 shows the morphological characteristics. Supplementary Table 1 shows the difference of VM and CSC marker expression of the TNBC and non-TNBC cases. PAS/CD31 double staining indicated that there was 66.7% with VM in the TNBC group, which was more than the 15.1% in the non-TNBC group (χ2= 5.270, P =0.020). Approximately 40.7% of the patients were positive for ALDH1 in the TNBC group, whereas 17.8% of the patients in the non-TNBC group were positive for ALDH1 (χ2= 6.381, P= 0.012). There were 37.1% of the TNBC cases expressing CD133, while 21.9% expressed CD133 in the non-TNBC group (χ2= 3.459, P = 0.043). At diagnosis, 33.3% and 19.2%of the TNBC and non-TNBC groups were CD44-positive and CD24-negative (χ2= 2.652, P = 0.090), respectively.
At the end of the follow-up on December 2008, a total of 71 patients survived. The mean survival of all patients was 86.2 ± 5.6 months. The Kaplan-Meier analysis indicated that the prognosis of the patients with VM was poorer than those without VM (Figure 2A, χ2= 5.907, P = 0.015). The survival of breast cancer patients with more CSCs was poorer than that of patients with fewer CSCs (Figure 2B-2D).
The relationship between VM and cancer stemlike cells in human breast cancer
Correlation analysis was used to detect the relationship between VM, TNBC type, and CSC marker expression in breast cancer. The results showed that VM was positively correlated with CSC markers ALDH1 and CD44/CD24(Table1). ALDH1 was positively correlated with TNBC type,VM, and VM-related proteins. CD44+/CD24- breast tumors were positively correlated with TNBC type, VM and HIF-1α.
The difference of ALDH1 and CD133 expression in TNBC MDA-MB-231 and non-TNBC MCF-7 cells
To investigate the difference of CSC marker expression in human breast cancer, TNBC MDA-MB-231 and non-TNBC MCF-7 cells were utilized to assess marker expression and VM formation in vitro. FACS indicated that there were more ALDH1+ and CD133+ populations in the TNBC MDA-MB-231 cells than in the MCF-7 cells (Figure 3A-3C). MDA-MB-231 cells expressed more ALDH1, CD133, and VM-related molecular VE-cadherin (Figure 3D and 3E). MDA-MB-231 cells were more likely to form VM-like channels on Matrigel than MCF-7 cells, and the cells lining VM-like channels were positive for CD133 and ALDH1 (Figure 3F and 3G).
Human cancer stem-like cells promote VM formation in human TNBC
To investigate the influence of CSCs on VM formation,ALDH1+ MDA-MB-231 cells, ALDH1- MDA-MB-231 cells,CD133+ MDA-MB-231 cells, and CD133- MDA-MB-231 cells were isolated to detect their ability to form VM. The ALDH1+ and CD133+ MDA-MB-231 cells formed stem-like cell spheres, while the ALDH1-negative or CD133-negative cells failed to form stem-like cell spheres (Figure 4A and Supplementary Figure S1A). Western blot showed that the ALDH1+ and CD133+ MDA-MB-231 cells expressed more VE-cadherin than the ALDH1-negative and CD133-negative MDA-MB-231 cells, and less E-cadherin was expressed in these cells (Figure 4B and Supplementary Figure S1B).Three-dimensional cell culture indicated that the ALDH1+and CD133+ MDA-MB-231 cells formed VM-like channels;however, the ALDH1-negative and CD133-negative MDAMB-231 cells lost the ability to form VM channels (Figure 4C, 4D and Supplementary Figure S1C, S1D). The ALDH1+and CD133+ MDA-MB-231 cells were more invasive and showed more migration ability than the controls (Figure 4 and Supplementary Figure S1).
Spatial relationship between the expression of stem cell markers and vasculogenic mimicry in breast cancer
In this study, fluorescein-labeled dextran was injected into the peripheral circulation of breast cancer-bearing mice via tail vein, which marked the blood flow. Then, whole mount staining was used to indicate endothelial cells labeled with red, breast cancer stem cells labeled with blue, and dextran labeled with green. The 3D reconstructed images indicated that the irregular blood flow without endothelium was due to VM channels that were connected to endothelium-dependent vessels. There were more VM vessels and CD133 or ALDH1-positive tumor cells in the MDA-MB-231 tumors than in the MCF-7 tumors (Figure 5A-5D). A 3D reconstructed image showed that the CSCs were closer to VM vessels than endothelium-dependent vessels (EDV) (Figure 5A-5D).

Figure 1 The morphological characteristics of human TNBC and non-TNBC. (A) H&E staining and IHC for ER, PR, and HER2 of human TNBC and non-TNBC. Tumor nests consist of poorly differentiated small tumor cells in TNBC, and necrosis is located in the center of a tumor nest(indicated by a black arrow). A number of tumor cells are undergoing mitosis. (B) Double staining for CD31 and PAS and IHC for VEcadherin, MMP2, and HIF-1α of human TNBC and non-TNBC. CD31/PAS double staining shows that TNBC has more VM channels compared with non-TNBC. The arrow indicates a VM channel that is formed by a PAS-positive matrix and tumor cells in TNBC. (C) IHC for ALDH1,CD44, CD24, and CD133 of human TNBC and non-TNBC. The scale bar = 100 μm.
Cancer stem-like cells promote VM formation in TNBC-bearing mice
TA2 breast cancer was derived from spontaneous breast
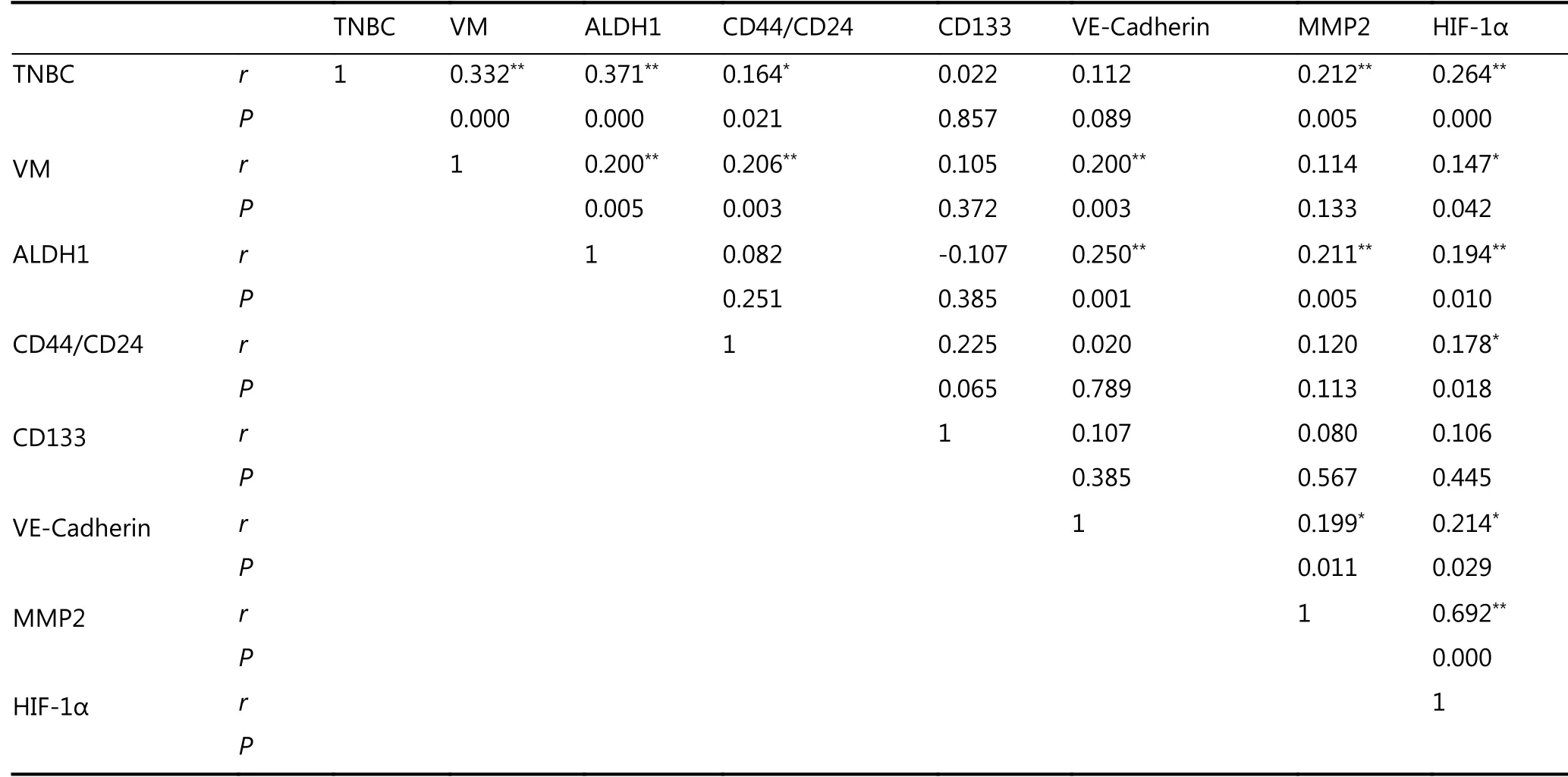
Table 1 The correlation of cancer stem-like cells proteins, VM, and VM-associated proteins
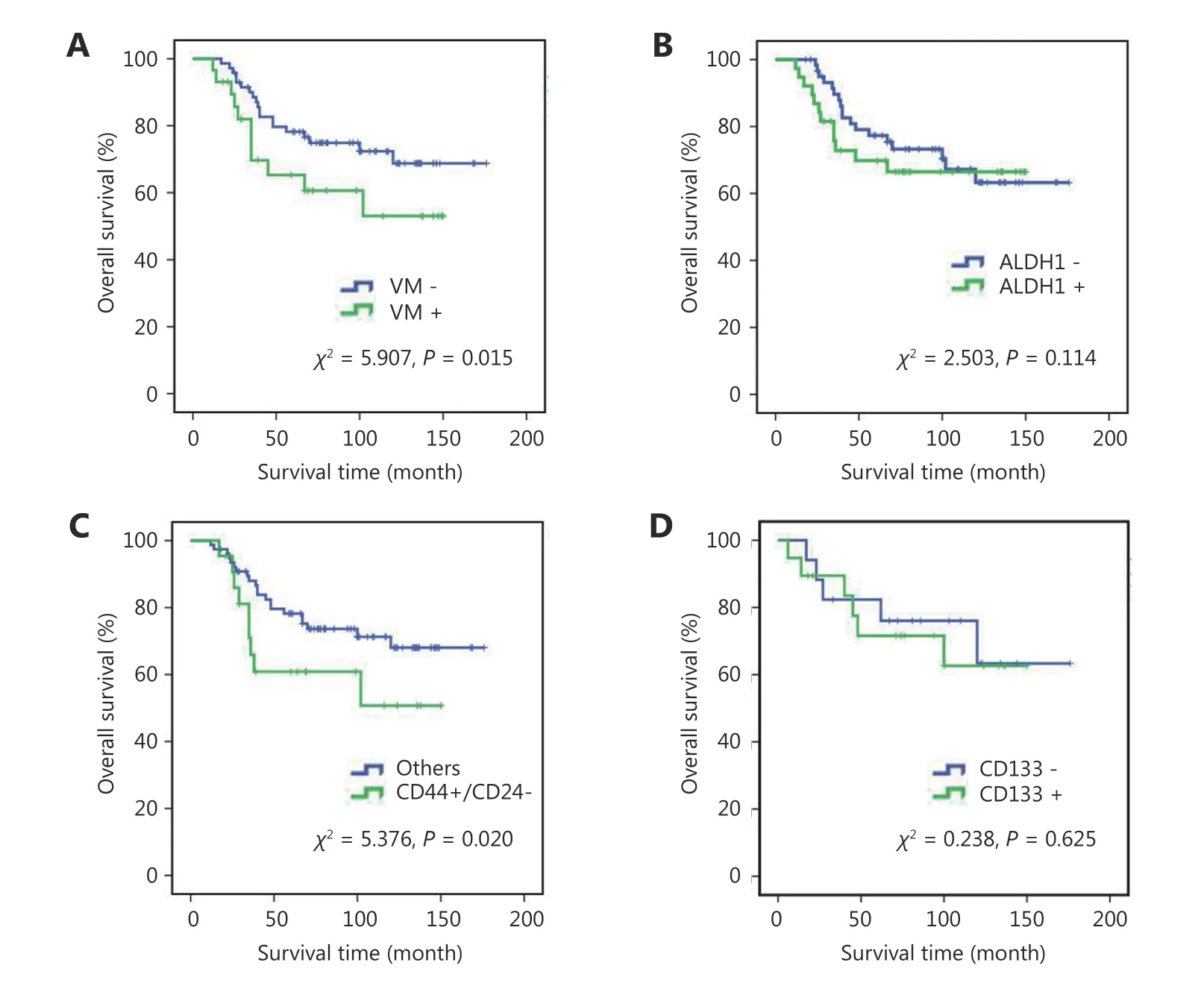
Figure 2 Kaplan-Meier survival analysis of human breast cancer. (A) Overall survival of breast cancer with VM and breast cancer without VM. (B) Overall survival of ALDH1+ and ALDH1- breast cancer. (C) Overall survival of CD44+/CD24- and the other breast cancers. (D) Overall survival of CD133+ and CD133- breast cancer.
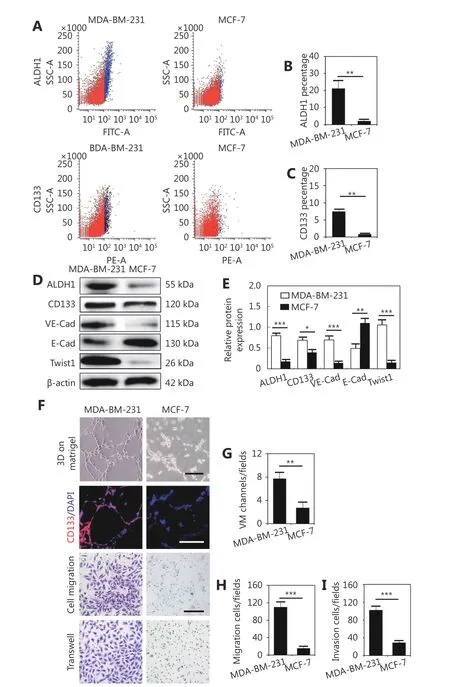
Figure 3 The difference of ALDH1 and CD133 expression in MDA-MB-231 and MCF-7 cells. (A) Representative FACS analyses of the ALDH1+ and CD133+ populations of MDA-MB-231 and MCF-7 cells. (B) Quantification of the ALDH1+ population in MDA-MB-231 and MCF-7 cells. (C) Quantification of the CD133+ population in MDA-MB-231 and MCF-7 cells. (D) Western blot showed the expression of ALDH1, CD133, VE-cadherin, E-cadherin, and Twist1 in MDA-MB-231 and MCF-7 cells. (E) Quantification of the expression of ALDH1,CD133, VE-cadherin, E-cadherin, and Twist1 in MDA-MB-231 and MCF-7 cells. (F) Matrigel cell culture shows that MDA-MB-231 cells formed more VM-like channels than MCF-7 cells on Matrigel. CD133 expressed in MDA-MB-231 cells lining VM. Cell migration and cell invasion analysis of MDA-MB-231 and MCF-7 cells. (G) Quantification of VM-like channels in MDA-MB-231 and MCF-7 cells. (H) Cell migration quantification of MDA-MB-231 and MCF-7 cells. (I) Cell invasion quantification of MDA-MB-231 and MCF-7 cells. The scale bar =100 μm, and the error bar indicates the SD (standard deviation). ** means P < 0.01, *** means P < 0.001.
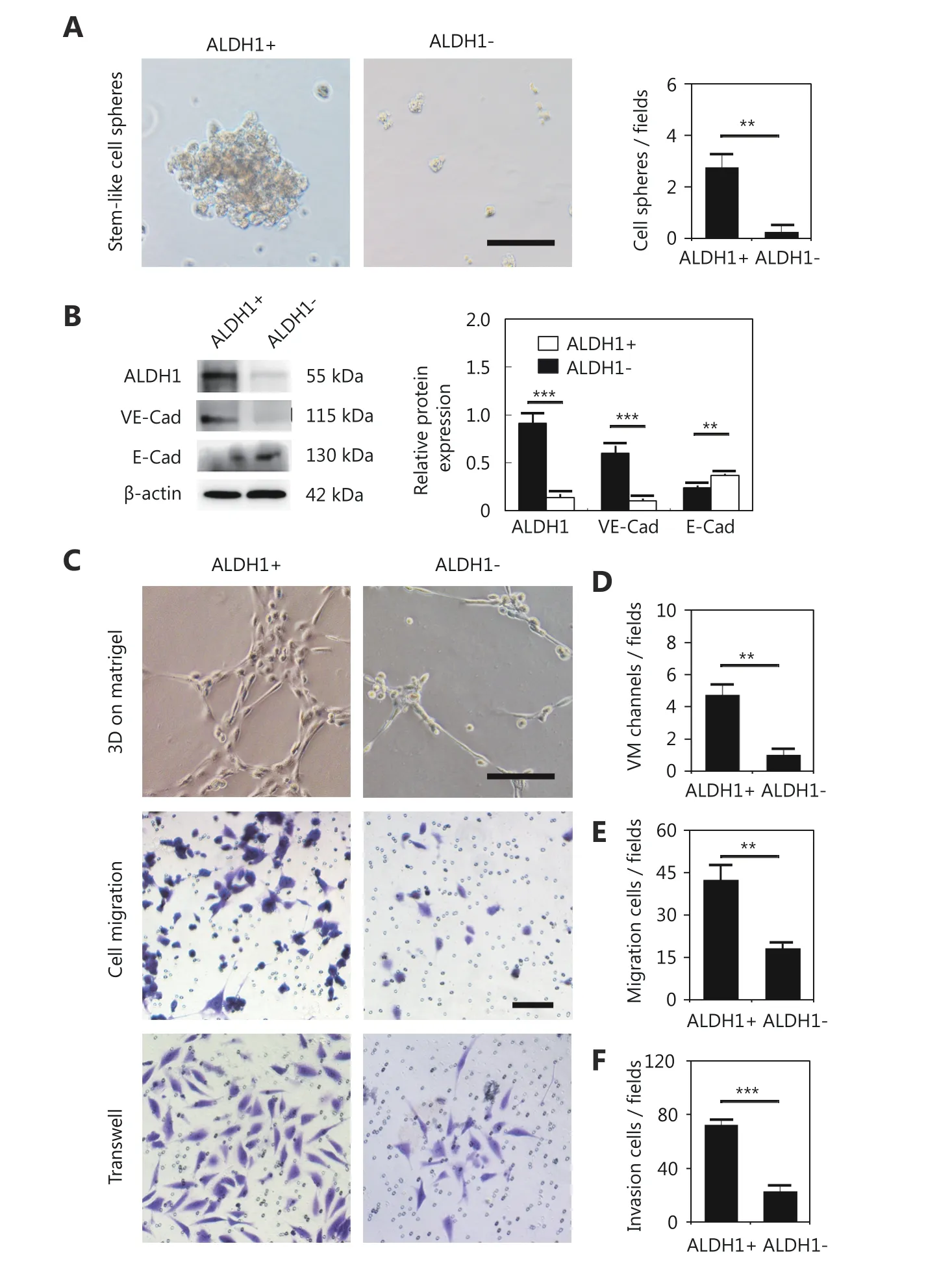
Figure 4 The difference between the ALDH1+ and ALDH1- MDA-MB-231 cells in sphere formation efficiency, VM-associated protein expression, VM-like channel formation, cell migration and cell invasion. (A) Stem-like cell sphere formation of the ALDH1+ and ALDH1-MDA-MB-231 cells. (B) Western blot detection of the expression of ALDH1, VE-cadherin, and E-cadherin in ALDH1+ and ALDH1- MDA-MB-231 cells. Quantification of the Western blot indicates that the ALDH1+ MDA-MB-231 cells expressed more ALDH1 and VE-cadherin than the ALDH1- MDA-MB-231 cells. (C) VM-like channel formation, cell migration and cell invasion of the ALDH1+ and ALDH1- MDA-MB-231 cells. (D) Quantification of VM-like channel formation of the ALDH1+ and ALDH1- MDA-MB-231 cells. (E) Quantification of cell migration of the ALDH1+ and ALDH1- MDA-MB-231 cells. (F) Quantification of cell invasion of the ALDH1+ and ALDH1- MDA-MB-231 cells. The scale bar = 100 μm, and the error bar indicates the SD (standard deviation). ** means P < 0.01, *** means P < 0.001.
cancer in TA2 mice. These tumors are triple-negative and easily develop metastatic sites in the lungs, liver, and spleen(Supplementary Figure S2A-S2G). The tumors expressed a high level of ALDH1 and CD133 (Supplementary Figure S3). To validate the relationship between CSCs and VM in a TNBC animal model, CSCs in TA2 breast cancer cells were isolated and engrafted into mice to detect VM formation and VM-related protein expression. The ratio of tumor formation and the tumor size in the ALDH1+ tumor cells were higher than those in the ALDH1- tumor cells (Figure 6A and 6C).Similar results were found in the CD133+ and CD133- tumor cells (Figure 6B and 6C). There were more VM channels in the ALDH1+ tumors than the ALDH- tumors, and the CD133+ tumors had more VM channels than the CD133-tumors (Figure 6D). The ALDH1+ tumors and the CD133+tumors expressed more ALDH1, CD133, VE-cadherin, and Twist1 than those in the ALDH1- tumors and the CD133-tumors (Figure 6E-6H).
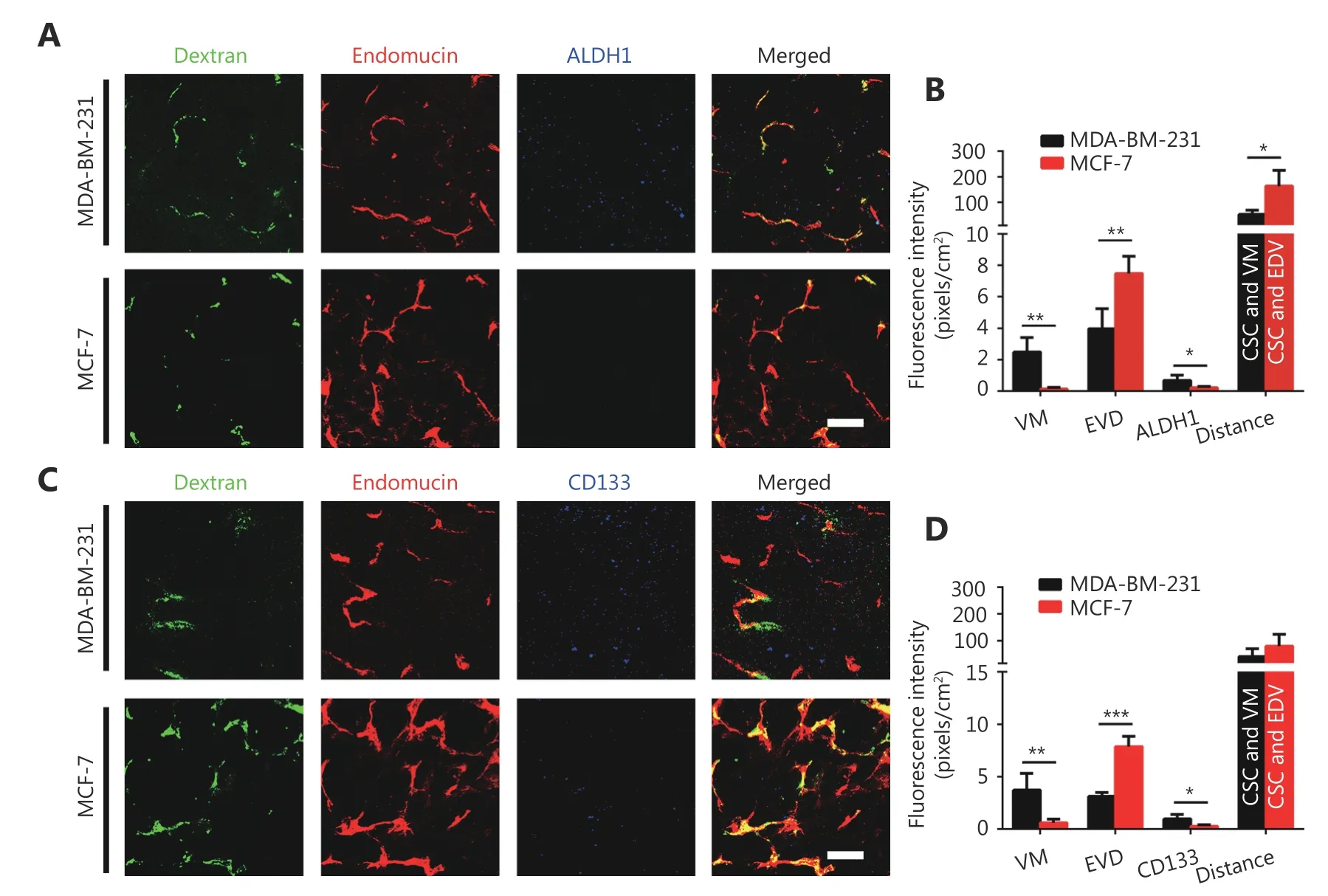
Figure 5 The spatial relationship between breast cancer stem-like cells and tumor microcirculation. (A) 3D image of dextran, EVD and ALDH1+ tumor cells. Laser confocal microscopy indicates that the irregular blood flow of uncoated endothelium is VM channels. (B)Quantification of VM, EVD, and ALDH1+ tumor cells in the MDA-MB-231 and the MCF-7 tumors and distance from ALDH1-positive tumor cells to blood vessels. (C) 3D image of dextran, EVD and CD133+ tumor cells. (D) Quantification of VM, EVD, and CD133+ tumor cells in the MDA-MB-231 and the MCF-7 tumors, and distance from CD133+ tumor cells to blood vessels. The scale bar = 100 μm, and the error bar indicates the standard deviation (SD). * means P < 0.05, ** means P < 0.01, *** means P < 0.001.
Discussion
VM is lined by tumor cells, and the molecular mechanisms of VM formation are independent from the mechanisms regulating EDV2,18. The traditional anti-angiogenic agents target the molecular signaling pathway that regulates endothelial cells4,19. Recently, sunitinib, an inhibitor of VEGF receptor tyrosine kinase activity, accelerated the recurrence and visceral metastasis of VM-positive TNBC in an animal model10. Hypoxia induced by inhibition of EDV vessels caused VM formation, which provided the blood supply instead of EDV vessels. VM is partly responsible for the resistance of anti-angiogenic treatment to TNBC10.Inhibition of VM will require understanding of the molecular mechanism of VM.
VM tissues are PAS+/CD31-/CD34- and do not respond to Ulex europaeus agglutinin-1 by immunohistochemistry20-21.Cells lining VM channels maintain some characteristics of the malignant tumor but also have some endothelial cell functions and phenotypes20,22. VM-initiating cells are of particular interest to investigators. The formation of VM in malignant tumors is similar to vasculogenesis in embryonic development23-25. Folberg et al. and Seftor et al. found that melanoma cell lines with VM expressed not only melanoma cell-specific genes but also multiple genes that regulate cell plasticity in embryogenesis13,24. CSCs were associated with VM formation in malignant tumors. CSC markers CD133 and CD44 were significantly correlated with VM existence in human renal cell carcinoma26. Hepatocellular carcinoma cells lining VM channels expressed SOX2 and OCT427. A neural precursor marker, nestin, and multiple stem cell markers,including CD133, Oct4, Nanog, and Notch1, were expressed in U87 glioma xenografts and human non-small lung carcinomas with VM28.
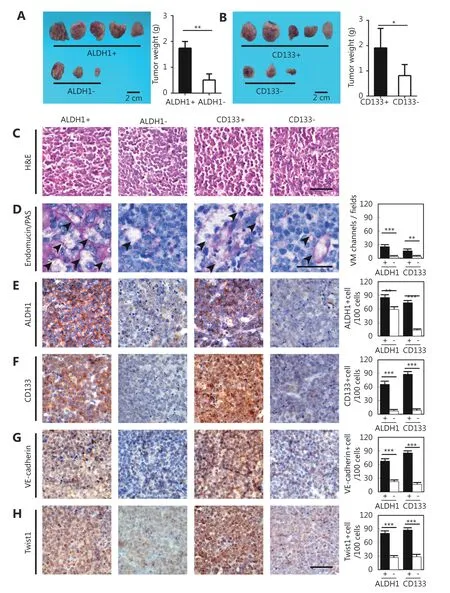
Figure 6 Effects of ALDH1+ and CD133+ tumor cells on TA2 breast tumor growth and VM-associated molecule expression. (A) Tumor size and tumor weight of ALDH1+ and CD133+ TA2 breast tumors. (B) H&E staining of ALDH1+ and CD133+ TA2 breast tumors. (C) Double staining for endomucin and PAS of ALDH1+ and CD133+ TA2 breast tumors. Quantification of VM number indicates that there are more VM channels in the ALDH1+ tumors than the ALDH- tumors, and the CD133+ tumors have more VM channels than the CD133- tumors. (E)IHC staining for ALDH1 in ALDH1+ and CD133+ TA2 breast tumors and quantification of ALDH1+ cells in different groups. (F) IHC staining for CD133 in ALDH1+ and CD133+ TA2 breast tumors and quantification of CD133+ cells in different groups. (G) IHC staining for VEcadherin in ALDH1+ and CD133+ TA2 breast tumors and quantification of VE-cadherin cells in different groups. (H) IHC staining for Twist1 in ALDH1+ and CD133+ TA2 breast tumors and quantification of Twist1-positive cells in different groups. The scale bar = 100 μm, and the error bar indicates the standard deviation (SD). * means P < 0.05, ** means P < 0.01, *** means P < 0.001.
In this study, CSC markers in breast cancer CD133,ALDH1, and CD44+/CD24- were positively correlated with VM and molecules involved in VM formation29. CSC markers were also related to TNBC subtype30-32. There were more tumors with VM in the TNBC group than in the non-TNBC group. TNBC cells express stem cell-specific markers and markers specific to mesenchymal stem cells6,33.Moreover, human TNBC cells MDA-MB-231 were enriched with ALDH1 and CD133 and had a higher capability to form VM than that of non-TNBC MCF-7 cells. Therefore, the results in this study confirmed that CSCs participated in VM formation.
Furthermore, ALDH1+ and CD133+ CSCs isolated from MDA-MB-231 and mouse TNBC cells formed VM channels in vitro and in vivo, while ALDH1-negative or CD133-negative breast cancer cells failed to form VM channels.MDA-MB-231 cells lining VM channels on Matrigel expressed CD133. Three-dimensional reconstructed images showed the spatial relationship between VM and CSCs. The CSCs were closer to VM vessels than to EDV vessels. These results directly indicated that tumor cells lining VM channels were derived from CSCs. Previous studies focused on the differentiation of CSCs into endothelium-like cells in vitro. In this study, three-dimensional reconstructed images showed the spatial relationship between VM and CSCs in an animal model. The results showed that the CSCs were closer to VM vessels than to EDV vessels. These results directly indicated that tumor cells lining VM channels were derived from CSCs.
CSCs can differentiate into multiple cell types34,35. Tumor generation and metastasis are based on CSC self-renewal and differentiation34-36. Many tumor endothelial cells have neoplastic origins37-40. Glioblastoma stem-like cells were successfully induced to differentiate into functional endothelial cells38. Glioblastoma and endothelial cells carry the same genomic alterations as tumor cells37. Tumors resistant to existing treatments have also been attributed to CSCs in malignant tumors41. CSCs are the source of tumor relapse and metastasis after treatment41. The differentiation and behavior of CSCs were the results of the responses of the tumor cells to the tumor microenvironment. For example,hypoxia caused by anti-angiogenic agents can contribute to CSC survival and metastatic potential42-43. Sunitinib, an antiangiogenic drug targeting the endothelium, caused more CD133+ subpopulation and VM formation in human TNBC grafts10. Hence, signaling pathways in CSCs and VM might be the target of anti-angiogenesis in malignant tumors.
There is more evidence that epithelial-mesenchymal transition (EMT) is an important promoter for VM formation in many malignant tumors2,44. The process of epithelial tumors transitioning to VM cells with mesenchymal features is similar to EMT in embryogenesis2,20.Many transcriptional factors involved in EMT, such as Twist1, Slug, bone morphogenetic protein 4 (BMP4), and Dickkopf-1 (DKK1), can induce VM formation6,27,45. Recent evidence revealed that EMT and CSCs have synergistic effects in VM promotion. Upregulation of high mobility group AThook 2 (HMGA2) resulted in gastric cancer cell VM and sphere formation46. The CSC markers CD44, ALDH1, Sox2,and Oct4 are also expressed at an increased level46. DKK1 also induced VM and the development of a CSC phenotype in non-small lung carcinoma6. Twist1 expression is related to TNBC tumors. There was a higher level of Twist1 in MDAMB-231 cells than in the MCF-710 cells. ADLH1+ and CD133+ TA2 breast cancer grafts expressed a high level of Twist1 and formed more VM channels. Inhibition of Twist1 disrupted the VM and reduced the CD133+ population in MDA-MB-231. Alternatively, upregulation of EMTregulating factors can also be induced by stem cell molecules.IMP3 and Sox2 induced Snail and Slug to promote EMT and metastasis in breast cancer47. Here, we showed that CD133+and ALDH1+ MDA-MB-231 cells expressed more Twist1 and VM-related VE-cadherin, which accelerates VM formation and tumor metastasis. This confirmed that there is some interaction between CSC differentiation and the EMT signaling pathway.
The Notch pathway is central to controlling cell fate both during angiogenesis and in CSCs from several tumors14,48.Notch4 is also involved in VM formation in melanoma and promoted CSC marker expression48-49. Notch4 regulates the expression of Nodal, a member of the TGF-β superfamily, by inducing the activity of an RBPJ-dependent pathway in an aggressive melanoma cell line49. The induction of Notch4 on Nodal is partly responsible for VM formation in melanoma.There are several Notch inhibitors, such as MK-0752 and N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine-tbutyl ester, which can reverse Notch4 effects in melanoma and pancreatic ductal adenocarcinoma cells50,51. Hence, they may be candidates for treatment of malignant tumors with VM.
In conclusion, CSCs lined VM channels directly.Additionally, CSCs provide more VM-related molecules to synergize VM formation. The signaling pathways that control CSC differentiation may also be potential therapeutic targets for TNBC.
Acknowledgments
This study was supported by the Student’s Platform for Innovation and Entrepreneurship Training Program, China(Grant No. 201510062001).
Conflict of interest statement
No potential conflicts of interests are disclosed.
 Cancer Biology & Medicine2019年2期
Cancer Biology & Medicine2019年2期
- Cancer Biology & Medicine的其它文章
- Factors associated with upstaging in patients preoperatively diagnosed with ductal carcinoma in situ by core needle biopsy
- Dorsomorphin induces cancer cell apoptosis and sensitizes cancer cells to HSP90 and proteasome inhibitors by reducing nuclear heat shock factor 1 levels
- Effects of palbociclib on oral squamous cell carcinoma and the role of PIK3CA in conferring resistance
- Hepatitis B virus X protein enhances hepatocarcinogenesis by depressing the targeting of NUSAP1 mRNA by miR-18b
- Identification of anticancer drugs to radiosensitise BRAFwild-type and mutant colorectal cancer
- Genetic polymorphisms and gastric cancer risk: a comprehensive review synopsis from meta-analysis and genome-wide association studies