
根癌农杆菌D-阿洛酮糖-3-差向异构酶基因克隆、结构预测及原核表达
2019-06-17 03:13朱星星杨培周杜明睿姜绍通
食品科学技术学报 2019年3期
朱星星, 杨培周, 杜明睿, 吴 芸, 姜绍通
(合肥工业大学 食品与生物工程学院/安徽省农产品精深加工省级实验室, 安徽 合肥 230009)
作为一种自然界天然存在但含量极少的糖,D-阿洛酮糖属于一种新型低热量甜味剂,其甜度相当蔗糖的70%,能量仅为蔗糖的0.3%[1-2],D-阿洛酮糖还具有特殊的生理活性,包括预防肥胖[2-3]、降血糖[3-4]、降血脂[5]、抗氧化[4]、保护神经[6]以及抑制癌细胞[7]等,因此,D-阿洛酮糖在降低糖吸收、调控身体机能以及抑制癌细胞等领域具有重要的应用价值,市场潜力广阔。
D-阿洛酮糖是D-果糖在C3位置的差向异构体,目前,主要采用化学合成和生物合成技术实现D-阿洛酮糖的生产。采用化学合成技术,钼酸盐在80~120 ℃能够将葡萄糖转化为D-阿洛酮糖[8];生物合成主要是通过D-塔格糖差向异构家族酶催化生成D-阿洛酮糖,与化学方法相比较,生物催化合成更为环保,分离纯化难度更小。通过基因工程技术,催化产D-阿洛酮糖的D-塔格糖差向异构家族酶已在大肠杆菌[9]、枯草芽孢杆菌[10]和酿酒酵母[11]中实现高效表达,基因来源主要为梭状芽孢杆菌[12-13],瘤胃球菌[9],根癌土壤杆菌[14-15],球形红菌[16],密螺旋体属菌[17]等。在国内,江南大学采用通过D-阿洛酮糖-3-差向异构酶基因克隆、工程菌构建和发酵控制等技术,将D-果糖转化为D-阿洛酮糖[17-18]。目前,全球D-阿洛酮糖的主要生产商韩国希杰第一制糖株式会社、英国泰莱和日本松谷集团都采用生物方法,将D-果糖转化为D-阿洛酮糖。因此,构建可催化产D-阿洛酮糖的基因工程菌是实现D-阿洛酮糖产业化的重要途径。
本研究根据NCBI公布的根癌农杆菌(Agrobacteriumtumefaciens)D-阿洛酮糖-3-差向异构酶(DPEase)基因的全序列,设计引物,扩增DPEase基因,通过生物信息学软件分析基因序列,预测翻译后的蛋白质结构和性质;构建原核重组质粒,检测DPEase基因在E.coliBL21中诱导表达,为进一步构建DPEase基因工程菌提供材料。
1 材料与方法
1.1 菌株与试剂
根癌农杆菌(Agrobacteriumtumefaciens)EHA105由唐晓凤博士馈赠,保存于合肥工业大学农产品精深加工实验室;E.coliDH5α、E.coliBL 21感受态细胞、FastPfuDNA聚合酶、pEASY-Blunt E1表达载体、氨苄青霉素、5-溴-4-氯-3-吲哚-β-D-半乳糖苷和异丙基-β-D-硫代吡喃半乳糖苷(IPTG)购自北京全式金生物公司;SanPrep柱式DNA胶回收试剂盒和SanPrep柱式DNA小量抽提试剂盒,生工(上海)公司;引物由生工(上海)公司合成。
1.2 仪器与设备
Nano- 100型分光光度计,杭州奥盛仪器公司;TC- 96型PCR仪,杭州博日公司;Universal Hood Ⅱ型全自动凝胶成像仪和蛋白电泳仪,美国Bio- Rad公司;EPS300 DNA型电泳仪和电泳槽,上海天能公司;E2695型高效液相色谱系统,美国Waters公司。
1.3 实验方法
1.3.1重组大肠杆菌的构建
根据NCBI数据库中公开的根癌农杆菌编码的DPEase基因 (KX098480.1),设计引物,上游引物:5′- CAGAAAAGCGAAAGAGACACC -3′;下游引物:5′- TGAGGATATTATCGCAAATC -3′;以提取的基因组DNA为模板,PCR扩增DPEase基因,回收目的片段,将目的片段与pEASY-Blunt E1载体连接,转化E.coli DH5α感受态细胞,挑取阳性克隆,提取质粒,测序,重组质粒转化E.coliBL21,通过抗性培养基筛选转化子,PCR鉴定拟转化子。
1.3.2生物信息学分析
采用NCBIBLAST比对核苷酸及氨基酸序列,通过NCBI在线ORF finder寻找基因编码框,利用Prot-Param在线软件分析蛋白质的理化性质,采用Signal P 4.1 Server预测蛋白信号肽,利用TMHMM 2.0 Server分析蛋白质的跨膜区,采用Predict Protein在线工具预测蛋白质二级结构,利用 Swiss-Model在线工具预测蛋白质三级结构。
1.3.3重组DPEase的诱导表达和分离纯化
重组E.coliBL21接种于200 mL含100 μg/mL氨苄的LB培养基中,37 ℃ 200 r/min振荡培养,过夜,然后加入终浓度为1 mmol/L的IPTG,在温度16 ℃和摇床转速150 r/min的条件下培养32 h,诱导重组菌表达外源蛋白。在4 ℃下,8 000 r/min离心10 min,收集菌体,生理盐水洗涤3次,缓冲液(50 mmol/L Tris-HCl,500 mmol/L NaCl,pH=8)重悬;在冰浴条件下,超声(功率300 W,工作3 s,间歇5 s,90次)破碎细胞,12 000 r/min离心15 min收集上清液,获得粗酶液;用0.22 μm微孔滤膜过滤粗酶液,将粗酶液加到层析柱上,低浓度咪唑洗脱液洗脱杂蛋白质,高浓度咪唑洗脱液洗脱目的蛋白质,目的蛋白质放入10 kDa的超滤管中浓缩除杂,SDS-PAGE检测重组DPEase。
层析柱预处理是在4 ℃条件下,采用上样缓冲液(50 mmol/L Na2HPO4,50 mmol/L NaH2PO4,500 mmol/L NaCl,pH=7.4)平衡Ni2+-Chelating Sepharose Fast Flow亲和层析柱。低浓度咪唑洗脱液为50 mmol/L Na2HPO4,50 mmol/L NaH2PO4,500 mmol/L NaCl,50 mmol/L咪唑,pH=7.4。高浓度咪唑洗脱液为50 mmol/L Na2HPO4,50 mmol/L NaH2PO4,500 mmol/L NaCl,500 mmol/L咪唑,pH=7.4。
1.3.4D-阿洛酮糖-3-差向异构酶活力测定
设置的5 mL催化反应体系中含有1 mL酶液、500 g/L D-果糖和50 mmol/L Tris-HCl(pH=8),在55 ℃和150 r/min条件下保温10 min,采用高效液相色谱法测定D-阿洛酮糖的含量[19],液相色谱柱为RezexTM RCM-monosaccharide Ca2+(Phenomenex IncTorrance, USA),检测器为2410型示差检测器,流动相为超纯水。设置参数:柱温80 ℃,流速0.4 mL/min,进样量10 μL。单位酶活力定义为1 min生成1 μmol D-阿洛酮糖为一个酶活力单位,以U/mL表示。
2 结果与分析
2.1 重组大肠杆菌的构建结果
以提取的根癌农杆菌基因组DNA为模板,根据DPEase序列设计的引物进行扩增,电泳结果见图1。

泳道M 为marker,1为阴性对照,2~4为扩增产物。图1 根癌农杆菌DPEase基因的PCR扩增结果Fig.1 PCR amplification of A. tumefaciens DPEase gene
由图1可知,电泳检测出一条约900 bp的目的片段,与预期大小一致。
目的片段与表达载体pEASY-Blunt E1连接,转化E.coliDH5α,通过抗性筛选,提取转化子质粒,测序验证正确,转化导入宿主菌E.coliBL21中,通过抗性固体培养基筛选转化子,采用DPEase引物PCR扩增转化子基因组,鉴定工程菌,见图2。
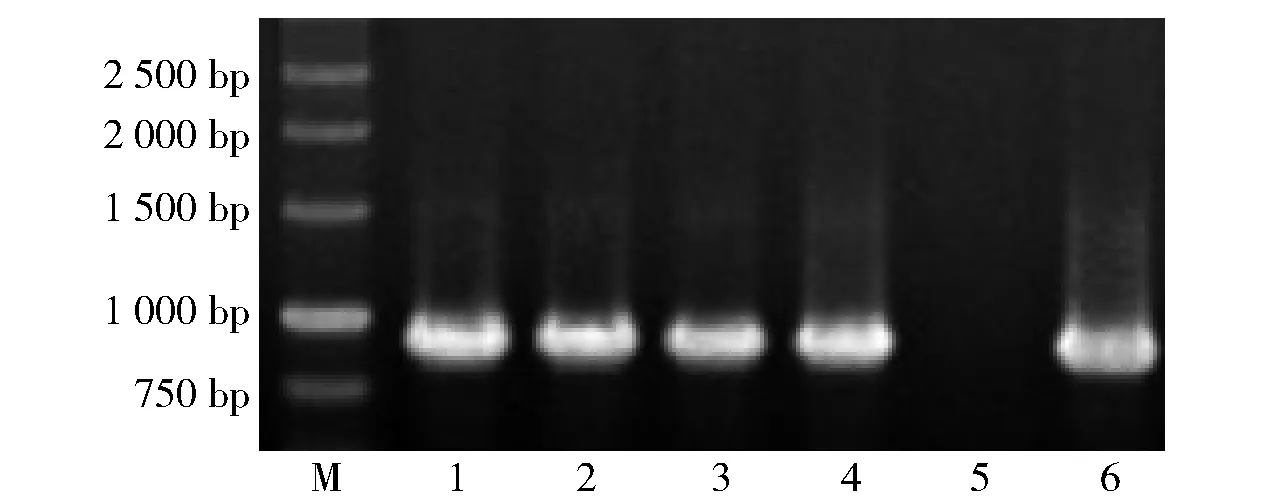
泳道M为marker,1~4为拟转化子扩增产物,5为阴性对照,6为阳性对照。图2 PCR鉴定DPEase E. coli BL21转化子Fig.2 PCR identification of DPEase transformants of E. coli BL21
由图2可知,被检测的转化子都能扩增出与目的基因预期一致的DNA条带,初步说明构建的表达质粒已转化入E.coliBL21细胞内。
2.2 生物信息学分析结果
2.2.1序列分析
对测定的DPEase序列进行开放阅读框分析,结果表明,该序列包含1个完整的开放阅读框,由870 bp核苷酸组成,编码289个氨基酸。通过在线同源性分析,该基因与NCBI的Genbank数据库中根癌农杆菌编码D-阿洛酮糖-3-差向异构酶基因 NCIM:2942(KX098480.1)的核苷酸序列相似性为99.43%。
2.2.2DPEase蛋白的理化性质分析
采用软件Prot Param预测DPEase的理化性质,结果显示,DPEase蛋白由289个氨基酸组成,其中含量最高是甘氨酸G,占11.07%,其次是丙氨酸A,占10.73%,最低的是谷氨酰胺Q和半胱氨酸C;带负电的残基(Asp+Glu)37个,带正电的残基(Arg+Lys)31个;其理论分子质量31 493.62,共由4 406个原子组成,分子式为C1 410H2 182N390O417S7,等电点PI为6;根据蛋白质的平均疏水性值判断蛋白质的疏水性,该蛋白为亲水性蛋白。
2.2.3信号肽和跨膜区的预测结果
通过Signal P 4.1 Server的人工神经网络算法对DPEase蛋白进行信号肽预测,结果如图3。
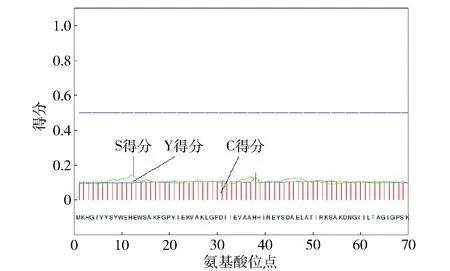
图3 Signal P- 4.1预测DPEase蛋白信号肽得分Fig.3 Prediction scores of DPEase signal peptide by Signal P- 4.1
由图3可知,Y得分最高分值(0.143)为第12位的组氨酸残基,S得分为0.107,DPEase基因所编码的蛋白无信号肽,为非分泌蛋白。
利用TMHMM 2.0 Server进行DPEase蛋白的跨膜区分析,结果见图4。

图4 基于TMHMM的后验概率跨膜区分析结果Fig.4 Analysis of transmembrane region based on TMHMM posterior probability
由图4可见,该蛋白质在膜内的概率仅为0.063 42,所编码的289个氨基酸从1到289全部在膜外;跨膜螺旋数为0,图中未出现矩形跨膜区。因此,该蛋白质位于细胞膜外,无跨膜区。
2.2.4DPEase蛋白的二级结构
Predict protein预测结果表明,DPEase蛋白含有13个α-螺旋和8个β-折叠,α-螺旋占38.41%,β-折叠47.06%,另外,14.53%的氨基酸处于无规则卷曲状态;非跨膜蛋白、α-螺旋跨膜蛋白和β-桶状跨膜蛋白的可能性依次为0.997 2、0.002 6和0.000 2,因此,DPEase属于非跨膜蛋白,预测结果与TMHMM 2.0 Server软件分析结果一致。
2.2.5DPEase蛋白的三级结构
根据Swiss-Model在线软件进行建模预测,采用D-阿洛酮糖-3-差向异构酶2hk1.1.A为模型建立DPEase蛋白的三级结构,结果见图5。

图5 DPEase蛋白的四聚体和单体结构Fig.5 Tetramer and monomer structure of DPEase
由图5可知,DPEase的氨基酸序列与2hk1.1.A模板的序列一致性为99.65%,建模的可信度较高。该DPEase的氨基酸序列在191位与模型有差异,此位点不属于活性结合位点;另外,I66、G67、G106、A107、W112、E150、L152、E156、D183、H186、H209、R215、E244和I257为D-果糖催化活性位点;E150、D183、H209和E244为Mn2+离子结合位点。
2.3 重组大肠杆菌的诱导表达结果
在16 ℃下,1 mmol /L IPTG 诱导重组菌E.coliBL21表达DPEase,结果见图6。

图6 IPTG 诱导E.coli BL21工程菌表达DPEase酶活力Fig.6 DPEase activities of E.coli BL21 engineered strain induced by IPTG
由图6可知,随着诱导时间增长,目的蛋白表达量增大,而当诱导24 h后,酶活增加缓慢,目的蛋白表达量在28 h时达到最大值0.32 g/L,酶活性最高为3.8 U/mL。
2.4 重组DPEase的分离纯化结果
通过Ni2+-Chelating Sepharose Fast Flow亲和层析柱,利用重组pEASY-Blunt E1-DPEase质粒带有的组氨酸标签和不同浓度的咪唑洗脱液,分离出目的蛋白,SDS-PAGE结果见图7。
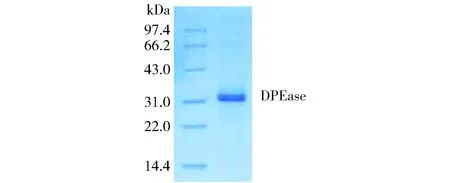
图7 SDS-PAGE分析纯化的DPEaseFig.7 SDS-PAGE analysis of purified DPEase
由图7可知,纯化后的蛋白质为单一条带,分子质量约为33 kDa,根癌农杆菌DPEase基因能够在E.coliBL21宿主中表达。
3 结 论
本文通过生物信息学分析根癌农杆菌DPEase蛋白结构,实验结果与已报道的结论接近。在分子质量预测上,本研究和Park等[20]研究表明,根癌农杆菌DPEase分子质量为33 kDa,与软件Prot Param预测的理论分子质量31 493.62相近;在DPEase晶体结构上,已有研究表明,DPEase蛋白为四聚体结构,每个单体呈典型TIM桶状,由(β/α)8结构组成,每个单体含有13个α-螺旋和8个β-折叠[14],与本研究通过生物信息学预测获得的结果相一致;在三级结构的活性位点分析方面,Kim等[14]通过定点突变研究表明,位点I66、A107、W112、E156和R215影响根癌农杆菌DPEase结合D-果糖的效率,H209影响Mn2+与底物结合[21],本研究通过预测,获得相似的结论。本研究通过分子克隆和原核表达检测根癌农杆菌DPEase的性质,与已知报道接近[14,21],研究表明,通过构建原核表达载体能够实现DPEase的高效表达,但其酶学性质以及在催化产D-阿洛酮糖等方面的应用还需进一步研究。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
肝博士(2022年3期)2022-06-30
海外星云(2021年9期)2021-10-14
食品与发酵工业(2020年16期)2020-01-07
农药科学与管理(2019年6期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
农药科学与管理(2019年8期)2019-11-23
江西医药(2018年8期)2018-10-24
中国糖料(2012年1期)2012-09-10
