
阿维巴坦类似物的合成及抗菌活性研究
2019-06-12 07:29叶海伟周丽萍陈云华
西北大学学报(自然科学版) 2019年3期
叶海伟,周丽萍,陈云华
(台州职业技术学院 化学制药研究所,浙江 台州 318000)
经典β-内酰胺抗生素的滥用易导致细菌的耐药,使人们对抗生素产生“恐惧”心理。近年来,新型β-内酰胺酶抑制剂的复方品种呈增长趋势,如阿维巴坦、Relebactam等新型巴坦类药物的问世无疑给处于低谷的抗生素领域吹来一股新风[1]。
2015年2月,由阿维巴坦与头孢他啶组成的复方产品Avycaz获得美国FDA批准上市,用于治疗成人复杂性腹腔内感染及尿路感染,与头孢洛林、氨曲南等其他复方产品分别处于临床Ⅱ和Ⅰ期中,是目前最被看好的非β-内酰胺类抑制剂[2-3]。阿维巴坦、Relebactam均为7-氧代-二氮杂二环[3.2.1]辛烷衍生物,两者具有相同的母核结构,仅是侧链酰胺键上取代基的不同,如图1所示。
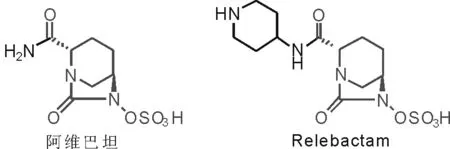
图1 阿维巴坦和Relebactam的结构Fig.1 Structure of avibactam and relebactam
另外,在传统的β-内酰胺类抑制剂中,他唑巴坦较舒巴坦多连接了一个三氮唑环,表现出毒性低、稳定性好、抑酶活性强等性质,而且与氨苄西林、阿莫西林、哌拉西林等抗生素联用均取得有效的协同作用[4]如图2所示。可见,三氮唑作为优秀的连接链模块被广泛应用于连接各种功能分子片段,得到的衍生物表现为更强劲的药效特点。
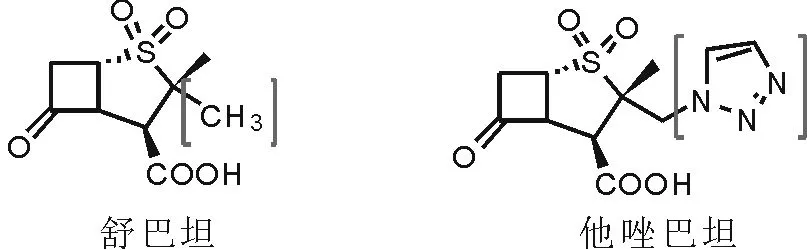
图2 舒巴坦和他唑巴坦的结构Fig.2 Structure of sulbactam and tazobactam
从氢键供体/受体性质上讲,1,2,3-三氮唑的C-4原子具有亲电性,该C—H键上的氢具有一定的氢键供体性质,且N-3的孤对电子又可以扮演氢键受体的角色;而1,4-二取代三氮唑可作为生物电子等排体,具有酰胺键的类似性质[5]。基于以上三氮唑的结构特点,查阅现有文献[6-9]对阿维巴坦类似物的结构修饰主要集中在酰胺键上连接不同的取代基,研究其对药物活性的影响,并无阿维巴坦母核结构与被取代的三氮唑通过亚甲基链接的衍生物报道。因而设计合成具有三氮唑结构的阿维巴坦类似物,并通过对铜绿假单胞菌和大肠杆菌最小抗菌浓度的测试,考察其抑菌活性大小,从而寻找活性更好的新颖巴坦类药物结构。
结合相关文献[4, 10-14],以(2S, 5R)-6-苄氧基-7-氧-1,6-二氮杂二环[3.2.1]辛烷-2-甲酸乙酯(2)为起始原料,经过还原和碘代合成(2S, 5R)-6-苄氧基-2-碘甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮(4),再与取代的三氮唑进行缩合使在二氮杂二环的亚甲基上引入三氮唑基团,经钯碳加氢脱除苄基,最后引入磺酸基,即可得到相应的类似物1,总收率为31%~43%(图3)。本路线具有反应路线简单,操作简便的优点。
1 实验部分
1.1 试剂与仪器
仪器:RY-1型熔点仪(天津市分析仪器厂)、Varian-400 MHz核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标,美国Varian公司)、Trace DSQ FINNIGSN质谱仪和Thermo IEC冷冻高速离心机(美国热电公司)、DZX-40BI 型立式自动电热压力蒸汽灭菌器(上海申安医疗器械厂)。
试剂:试剂均为AR,溶剂使用前均按标准方法处理。Mueller-Hinton肉汤(北京陆桥技术有限责任公司)、硫酸庆大霉素(山东鲁抗医药股份有限公司)、阿维巴坦(自制)。
1.2 实验方法
1.2.1 (2S 5R)-6-苄氧基-2-羟甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮(3)的合成 取250 mL四口烧瓶,加入原料(2S,5R)-6-苄氧基-7-氧-1,6-二氮杂二环[3.2.1]辛烷-2-甲酸乙酯(2, 25.0 g, 82.2 mmol),甲醇180 mL,降温至-5~-10℃,然后分批加入硼氢化锂(5.4 g, 246.6 mmol),并控制温度在0℃以下继续搅拌反应5 h后,加入饱和磷酸二氢钠100 mL进行淬灭反应,减压回收甲醇后,向剩余物中加入二氯甲烷进行萃取三次(80 mL×3),合并有机相,加入无水硫酸钠干燥,过滤,滤液进行减压浓缩后经柱层析纯化(流动相为正己烷/乙酸乙酯=2∶1(体积比)),得白色固体的(2S,5R)-6-苄氧基-2-羟甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮(3)18.2 g,收率为84%。Mp:233℃~235℃;1H-NMR (400 MHz, CDCl3)δ: 7.42-7.34 (m, 5H), 5.07 (d,J=12.0 Hz, 1H), 4.96 (d,J=12.0 Hz, 1H), 3.73-3.69 (m, 1H), 3.61-3.58 (m, 2H), 3.33-3.30 (m, 1H), 3.10 (d,J=12.0 Hz, 1H), 2.91 (m, 1H), 2.07-1.99 (m, 2H), 1.58-1.53 (m, 1H), 1.42-1.26 (m, 1H); MS (ESI):m/z(%) 263.1 [M+H]+。
1.2.2 (2S,5R)-6-苄氧基-2-碘甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮(4)的合成 在250 mL四口瓶中,加入(2S, 5R)-6-苄氧基-2-羟甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮(3, 7.9 g, 30.0 mmol)和二氯甲烷80mL,在冰浴下加入三苯基膦(9.4g, 36.0 mmol)、咪唑(3.1 g, 45.0 mmol)和碘(9.1 g, 36.0 mmol),搅拌下逐渐升至室温反应3h,加入饱和亚硫酸钠80 mL淬灭,用乙酸乙酯萃取(80 mL×3),合并有机相,无水硫酸钠干燥,过滤,滤液进行减压浓缩后经柱层析纯化(正己烷/乙酸乙酯=5∶1(体积比)),得浅黄色油状产物(2S,5R)-6-苄氧基-2-碘甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮9.7g,收率为87%。1H NMR (400 MHz, CDCl3)δ: 7.39-7.29 (m, 5H), 5.01 (d,J=12.0 Hz, 1H), 4.91 (d,J=12.0 Hz, 1H), 3.68-3.50 (m, 3H), 3.33-3.26 (m, 1H), 3.02-2.89 (m, 2H), 2.07-1.98 (m, 2H), 1.56-1.50 (m, 1H), 1.41-1.28 (m, 1H);MS(ESI):m/z(%) 395.0 [M+Na]+。

图3 阿维巴坦类似物的合成路线Fig.3 Route of avibactam analogues synthesis
1.2.3 阿维巴坦类似物1a的合成 在100 mL三口瓶中,加入(2S,5R)-6-苄氧基-2-碘甲基-1,6-二氮杂二环[3.2.1]辛烷-7-酮(4, 2.4 g),1H-1,2,3-三氮唑(4.8 g),碳酸氢钠(1.0 g),丙酮30 mL和水6 mL,室温下搅拌反应,TLC跟踪检测反应,减压蒸除丙酮,向剩余物中加入45mL乙酸乙酯进行萃取,分出有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,滤液进行减压浓缩后得到相应的中间产物,不经纯化,直接投入下一步。
在另一只100 mL三口瓶中,加入上述得到的相应中间产物(2S,5R)-2-((1H-1,2,3-三氮唑-1-)甲基)-6-苄氧基-1,6-二氮杂二环[3.2.1]辛烷-7-酮、10%钯碳0.3 g和乙醇20 mL,在氢气氛围下搅拌进行脱除苄基反应,反应过程采用TLC跟踪检测反应,反应结束后,加入硅藻土进行过滤后,滤液进行减压浓缩得到粗品,再加入吡啶15 mL和三氧化硫吡啶络合物(5.6 g, 35.0 mmol),室温下搅拌进行磺化反应8h后,减压浓缩反应液,再经柱层析纯化(V(二氯甲烷)∶V(乙酸乙酯)∶V(乙醇)=5∶4∶1),得到浅黄色油状物为阿维巴坦类似物1a为1.4 g,收率53%。1H-NMR(400MHz, CDCl3)δ:7.75(d,J=6.0Hz, 2H), 4.00-3.88 (m,2H), 3.73-3.69 (m, 1H), 3.61-3.58 (m, 2H), 3.33-3.21 (m, 1H), 2.91-2.88 (m, 1H), 2.07-1.99 (m, 2H), 1.42-1.26 (m, 1H)。MS (ESI):m/z(%) 304.1 [M+H]+。
根据上步类似的操作,合成阿维巴坦类似物1b~1j。
1b:淡黄色油状物,收率58%。1H-NMR (400 MHz, CDCl3)δ:7.88 (s,1H), 7.83 (s, 1H), 4.05-3.93 (m, 2H), 3.73-3.69 (m, 1H), 3.60-3.58 (m, 2H), 3.33-3.21 (m, 1H), 2.91-2.88 (m, 1H), 2.07-1.99 (m, 2H), 1.43-1.28 (m, 1H)。MS (ESI):m/z(%) 304.0 [M+H]+。
1c:黄色油状物,收率52%。1H-NMR (400 MHz, CDCl3)δ: 7.81 (s, 1H), 4.00-3.88 (m, 2H), 3.73-3.68 (m, 1H), 3.60-3.55 (m, 2H), 3.36-3.25 (m, 1H), 2.90-2.88 (m, 1H), 2.36 (s, 3H), 2.07-1.99 (m, 2H), 1.42-1.26 (m, 1H); MS (ESI):m/z(%) 318.1 [M+H]+。
1d:浅黄色油状物,收率45%。1H-NMR (400 MHz, CDCl3)δ: 7.89 (s, 1H), 4.00-3.88 (m, 2H), 3.75-3.69 (m, 1H), 3.61-3.58 (m, 2H), 3.33-3.21 (m, 1H), 2.91-2.88 (m, 1H), 2.26 (s, 3H), 2.13-2.05 (m, 2H), 1.41-1.28 (m, 1H); MS (ESI):m/z(%) 318.3 [M+H]+。
1e:黄色黏状物,收率59%。1H-NMR (400 MHz, CDCl3)δ: 8.21 (s, 1H), 4.10-3.95 (m, 2H), 3.75-3.69 (m, 1H), 3.61-3.58 (m, 2H), 3.35-3.27 (m, 1H), 2.91-2.88 (m, 1H), 2.25-2.19 (m, 1H), 1.77-1.70 (m,1H), 1.4-1.26(m, 1H); MS(ESI):m/z(%) 338.1 [M+H]+。
1f:黄色油状物,收率48%。1H-NMR (400MHz, CDCl3)δ: 8.18 (s, 1H), 4.15-3.98 (m, 2H), 3.75-3.67 (m, 1H), 3.61-3.58 (m, 2H), 3.35-3.27 (m, 1H), 2.91-2.85 (m, 1H), 2.26-2.19 (m, 1H), 1.78-1.70 (m, 1H), 1.41-1.32 (m, 1H); MS (ESI):m/z(%) 381.9 [M+H]+。
1g:米黄色油状物,收率45%。1H-NMR(400MHz, CDCl3)δ: 4.18-4.09 (m,2H), 4.01-3.95 (m, 1H), 3.75-3.68 (m, 1H), 3.63-3.58 (m, 2H), 3.36-3.29 (m, 1H), 2.29-2.22 (m, 1H), 1.78-1.70 (m, 1H), 1.43-1.34 (m, 1H); MS (ESI):m/z(%) 459.9 [M+H]+。
1h:黄色油状物,收率49%。1H-NMR(400MHz, CDCl3)δ: 8.13 (s, 1H), 4.00-3.89 (m, 2H), 3.71-3.65 (m, 1H), 3.60-3.53 (m, 2H), 3.36-3.25 (m, 1H), 2.91-2.88 (m, 1H), 2.07-1.99 (m, 2H), 1.38-1.25 (m, 1H); MS (ESI):m/z(%) 349.1 [M+H]+。
1i:黄色油状物,收率45%。1H-NMR (400MHz, CDCl3)δ: 7.41 (s, 1H), 4.09-3.98 (m, 1H), 3.90-3.81 (m, 1H), 3.73-3.68 (m, 1H), 3.65-3.56 (m, 2H), 3.36-3.25 (m, 1H), 2.93-2.89 (m, 1H), 2.07-2.00 (m, 2H), 1.38-1.25 (m, 1H); MS (ESI):m/z(%) 372.2 [M+H]+。
1j:淡黄色油状物,收率43%。1H-NMR(400MHz, CDCl3)δ: 7.93 (s, 1H), 4.15-4.08 (m, 2H), 3.78-3.69 (m, 1H), 3.63-3.57 (m, 2H), 3.33-3.21 (m, 1H), 2.77-2.64 (m, 1H), 2.13-2.07 (m, 2H), 1.41-1.29 (m, 1H); MS (ESI):m/z(%) 372.1 [M+H]+。
1.2.4 抗菌活性实验 参照美国临床实验室标准化委员会(NCCLS)最小抑菌浓度(MIC)的测定方法[15],以阿维巴坦(avibactam)和硫酸庆大霉素(gentamicin sulfate)为阳性对照,测试了目标化合物对铜绿假单胞菌(pseudomonas aeruginosa, P. a)和大肠杆菌(escherichia coli, E. c.)进行体外抗菌性能的测试。
具体采用常规的测试方法如下:
1)用接种环挑取菌落3~5个,接种于4~5mL的M肉汤中,35℃孵育2~6h,增菌后的对数生长期菌液用MH肉汤校正浓度,约含1×108~2×108CFU/mL。
2)取无菌96孔板,B~H孔各加MH肉汤培养基99μL;A孔加MH肉汤培养基199μL和最适稀释度的受试化合物溶液1μL。B~H孔分别加入最适稀释度倍比稀释的浓度梯度药物各1μL,最后每孔加测试细菌100 μL,各孔中DMSO含量均不超过0.5%(体积分数);阳性药物按一定浓度梯度进行稀释后每孔加100μL再加测试细菌100μL。各药敏板于35℃培养。
3)将接种好的药敏板,置35℃普通空气孵箱中孵育16~20 h,用肉眼观察每孔的浑浊度。以在小孔内完全抑制细菌生长的最低药物浓度为MIC。
2 实验结果和讨论
2.1 合成的优化
以阿维巴坦关键中间体(2S, 5R)-6-苄氧基-7-氧-1,6-二氮杂二环[3.2.1]辛烷-2-甲酸乙酯(2)为起始原料,在醇体系下,经硼氢化锂还原,即得化合物3,收率为84%。羟基的碘代考虑底物7-氧代-二氮杂二环[3.2.1]辛烷的结构,反应条件太苛刻容易氧化变质,选择较为温和的三苯基膦/咪唑/碘体系,合成中间体4,收率为87%。
接着,借助碘原子的易离去性,4与取代的三氮唑进行缩合反应使在二氮杂二环的亚甲基上引入三氮唑基团,经钯碳加氢脱除苄基,按文献方法[13]引入磺酸基,即可得到相应的类似物1,收率为31%~43%不等。以铜绿假单胞菌和大肠杆菌为测试对象,对相应化合物进行抗菌活性的检测。
2.2 目标化合物的活性研究
由表1可知通过对7-氧代-二氮杂二环[3.2.1]辛烷的结构进行研究发现,采用三氮唑或取代的三氮唑代替阿维巴坦结构相应位置上的酰胺键,并通过亚甲基进行键合来进行修饰,通过对铜绿假单胞菌和大肠杆菌进行体外抗菌性能的测试来筛选相应的衍生物活性,发现三氮唑有吸电子基团(-Br,-NO2或-CF3)取代,达到较好的的抗菌活性(类似物1g~1j),类似物1i的抗菌活性甚至要远好于阿维巴坦。这部分化合物我们将做进一步的药理、毒理等相关研究,具有良好的应用前景。
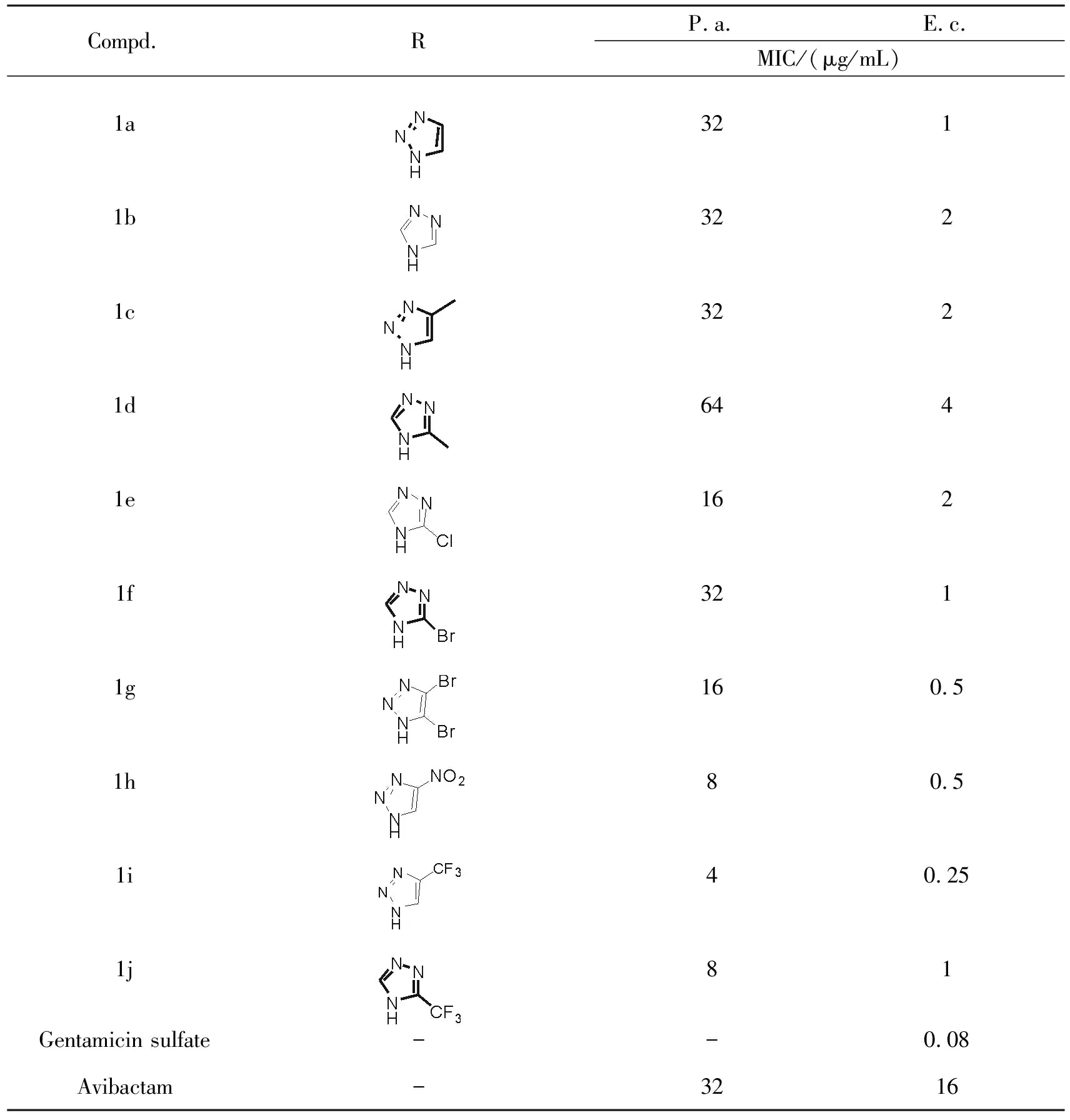
表1 化合物对相应细菌的抑制作用(MIC μg/mL)Tab.1 In vitro antimicrobial activities for target compounds expressed as MIC(μg/mL)
3 结 论
以阿维巴坦为先导化合物,对酰胺键及相关结构进行三氮唑的取代修饰,合成衍生物,通过相关的抗菌活性实验表明对革兰氏阳性菌(铜绿假单胞菌)和革兰氏阴性菌(大肠杆菌)具有良好的抗菌活性,对开发新型的巴坦类药物具有重要的意义,其他位置上的结构修饰和构效关系正在进一步研究中。
猜你喜欢
中华养生保健(2020年1期)2020-11-16
小读者(2020年2期)2020-03-12
作文成功之路·小学版(2020年11期)2020-02-01
下一代英才(酷炫少年)(2019年3期)2019-03-25
铜仁学院学报(2018年6期)2018-07-05
西北药学杂志(2018年4期)2018-02-12
作文周刊·高一版(2017年39期)2017-11-11
赤子(2017年4期)2017-06-30
中国洗涤用品工业(2016年2期)2016-02-28
中国继续医学教育(2015年5期)2016-01-07
