
气相色谱法分析丙烯腈反应器流出物中丙烯腈、乙腈、丙烯醛、丙烯酸
2019-06-03 07:54:56吴泽学
安徽化工 2019年2期
吴泽学
(中国石油化工股份有限公司安庆分公司检验计量中心,安徽安庆246002)
工业用丙烯腈主要用于生产腈纶纤维、ABS 塑料和SAN 树脂[1]。中石化安庆分公司丙烯腈装置采用Sohio公司丙烯氨氧化先进工艺技术[2-4],国内采用此技术的丙烯腈生产装置共有13 套。丙烯腈反应器流出物的组成分析是计算丙烯腈收率、丙烯转化率,评价催化剂性能的重要依据。丙烯腈反应器流出物的组成包括丙烯腈、乙腈、丙烯醛、丙烯酸、吡啶、反丁烯二腈、丙烯酰胺、丁二腈、一氧化碳、二氧化碳、丙烯、丙烷、氧气等有机和无机杂质[5]。现有丙烯腈反应器流出物中丙烯腈、乙腈、丙烯醛、丙烯酸的分析方法为使用两台色谱仪,一台装有PQ(80~100 目)2 m 长不锈钢柱用于分析丙烯腈、乙腈、丙烯醛,另一台装有PQS(80~100 目)1 m 长玻璃柱用于分析丙烯酸。国内也有使用Carbowax 20 M涂装4%Carbopack D-BA 的玻璃填充柱,采用程序升温来分析的方法[6],这类方法的缺点是使用一段时间后保留值改变,分离度下降且分析时间较长;乙腈与丙烯醛分离效果不好;而且需要两次进样及外标法定量,然而进样量及色谱条件如温度、流量的变化,容易影响样品分析结果的准确度。本方法为采用一台气相色谱,选用DB-FFAP(60 m×0.32 mm×0.50 μm)毛细管色谱柱,柱流量3.0 mL/min,分流比50∶1,采用程序升温及内标法定量,实现了一次进样同时分析丙烯腈反应器流出物中丙烯腈、乙腈、丙烯醛、丙烯酸,克服了现有分析方法需要两台气相色谱才能分别检测流出物中丙烯腈、乙腈、丙烯醛、丙烯酸的不足。本方法不仅缩短分析时间至20 min,而且明显改善了乙腈与丙烯醛的分离效果,减少了丙烯酸的吸附,各组分相对误差在-1.55%~+0.88%之间,满足了生产实际要求。
1 实验部分
1.1 材料与试剂
岛津GC-2014 气相色谱(具SPL 分流装置、氢火焰检测器);美国Thermo Atlas 8.1 色谱工作站;梅特勒托利多ME-204 电子天平;10 μL 注射器;50 mL 容量瓶。
丙烯腈(99.0%,国药集团化学试剂有限公司);丙烯醛(90%,SIGMA-ALDRICH);2- 丁酮(99.5%,国药集团化学试剂有限公司);乙腈(99.8%,国药集团化学试剂有限公司);丙烯酸(99%,东京化成工业株式会社);甲酸(99.5%,国药集团化学试剂有限公司)。
1.2 标样的配制
取一洁净、干燥恒重的50 mL 容量瓶,根据样品实际浓度配制浓度近似的反应器流出物标样,依次加入约50 mL 二级蒸馏水、240 μL 色谱纯丙烯腈、10 μL 色谱纯乙腈、7 μL 色谱纯丙烯酸、5 μL 色谱纯丙烯醛、40 μL 色谱纯2- 丁酮,2- 丁酮作为内标物[7],并分别在电子天平上准确称量,混匀,在室温下放置10 min。标样配制结果见表1。
1.3 色谱条件
色谱柱DB-FFAP(60 m×0.32 mm×0.50 μm)毛细管色谱柱,柱流量3.0 mL/min,分流比50∶1,量程×1,柱初始温度54℃,保持时间8 min,升温速率30℃/min,终温200℃,保持时间8 min,进样器温度230℃,检测器(FID)温度250℃,氢气压力60 kPa,空气压力50 kPa,MAKE UP 压力75 kPa。
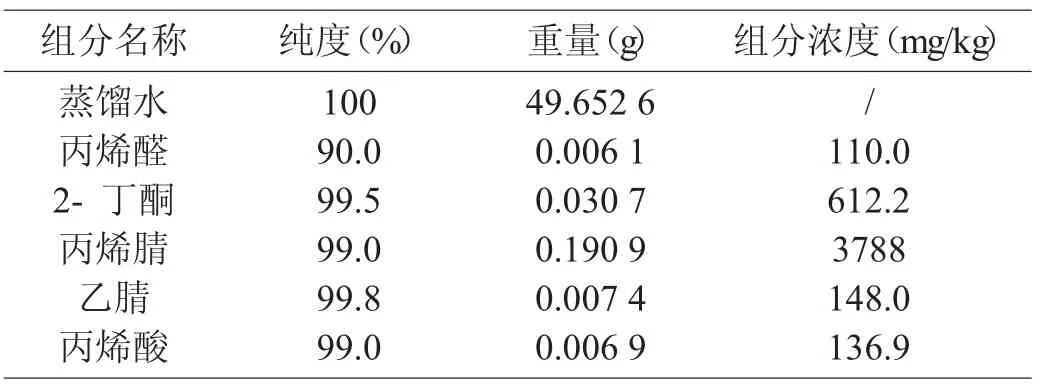
表1 丙烯腈反应器流出物标样配制明细表
1.4 分析步骤及定量方法
对现配制的丙烯腈反应器流出物标样进样,每次2 μL,共进五次标样,采用内标法定量[8],根据公式计算出丙烯醛、丙烯腈、乙腈、丙烯酸的相对质量校正因子,求出其平均值,结果见表2。
由于丙烯腈具有不稳定的双键结构,极易发生自聚、共聚、水解等[9],丙烯醛、丙烯酸极不稳定,可发生聚合反应,丙烯酸在FFAP 柱上有轻微吸附,故在实际分析过程中,应严格控制采样量及样品吸收温度,采样后应及时分析,分析前进2 至3 针丙烯腈反应器流出物样品,以减少毛细管柱对丙烯酸的吸附。

表2 各组分的相对质量校正因子
2 结果与讨论
2.1 色谱条件的优化
在色谱操作条件中,柱温、载气流速、分流比、进样量的选择对色谱组分的分离及分析时间有很大影响,色谱柱的选择与待测组分的分离和保留时间等密切相关,定量方法的选择则直接影响样品分析的准确度。
2.1.1 色谱柱选择
乙腈、丙烯酸的极性强,丙烯酸的沸点高,采用色谱法分离时,色谱柱选择不恰当,会导致色谱峰保留时间过长,色谱峰拖尾及峰型不对称等现象。通过对弱极性色谱柱HP-5(30 m×0.32 mm×0.25 μm)和中等极性色谱柱DB-FFAP(60 m×0.32 mm×0.50 μm)的实验比较,HP-5 色谱柱柱分析时间短,丙烯酸组分峰对称、拖尾小,但分离效果差,相比较而言,DB-FFAP 色谱柱操作条件范围广,分离效果好,但丙烯酸有轻微拖尾现象,故选择DB-FFAP 色谱柱较恰当。
通过实验反复比较,本方法色谱条件(见1.3)对丙烯腈反应器流出物丙烯腈、乙腈、丙烯醛、丙烯酸的分离有较好的效果,采用内标法定量,分析结果准确度高。
2.1.2 色谱柱老化
使用前将毛细管柱的头尾各裁掉5~10 cm,进样口端与色谱仪的进样口相连,毛细管柱另一端不与检测器连接,通入载气,设置载气柱流量为1.0 mL/min,依次将柱箱温度设置为100℃、180℃、220℃各老化1 h。分段老化结束后,将色谱参数设置到正常操作条件(见1.3),将毛细管柱另一端与检测器连接,注入7 针2 μL 现配的0.1%甲酸溶液,每次进样间隔10 min,这样做一方面减少丙烯酸色谱峰拖尾,另一方面使毛细管柱保持在酸性环境中,以减少毛细管柱对丙烯酸的轻微吸附作用。
2.1.3 定量方法优化
本方法采用内标法代替了外标法定量,克服了色谱条件中诸如柱温、载气流速、进样量等的变化对样品分析结果准确度的影响。
2.2 与现有方法比较
丙烯腈反应器流出物标样现有分析方法色谱图分别见图1、图2,采用DB-FFAP 毛细管柱,分析丙烯腈反应器流出物标样色谱图见图3。由图1、图2、图3 可知,本实验方法较现有分析方法节约了分析时间,由45 min减少到20 min,峰型更对称,减小了丙烯腈、丙烯酸的峰型扩展,改善了乙腈与丙烯醛的分离效果。由于采用一次进样代替原来的两次进样,内标法代替外标法定量,减小了样品的分析误差。
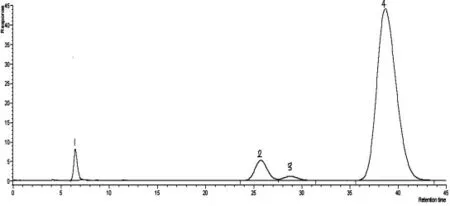
图1 PQ(80~100 目)填充柱分析丙烯腈反应器流出物标样色谱图
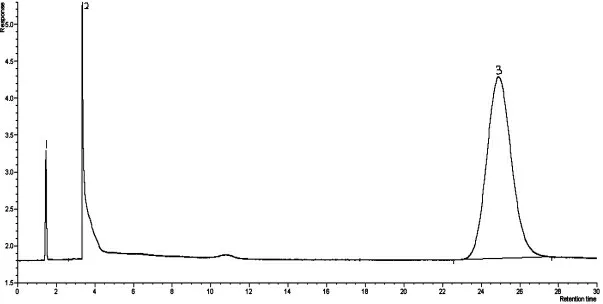
图2 PQS(80~100 目)玻璃柱分析丙烯腈反应器流出物标样色谱图
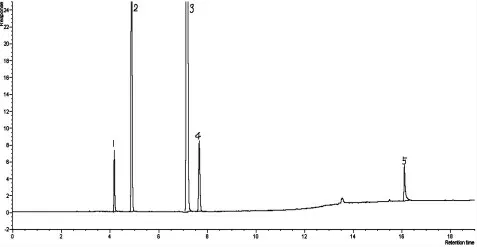
图3 DB-FFAP 毛细管柱分析丙烯腈反应器流出物标样色谱图
2.3 准确度与精密度试验
对现配制的丙烯腈流出物标样进样,每次2 μL,共进五次标样,采用内标法定量,计算出标样各组分的相对误差和相对标准偏差RSD[10],结果见表4。由表4 可知,采用毛细管色谱法,样品各组分相对误差为-1.55%~+0.88%,回收率为98.5%~100.9%,相对误差均小于5%,准确度良好。从测定结果看,样品相对标准偏差均小于2%。本方法有较好的重复性,可满足实际分析要求。
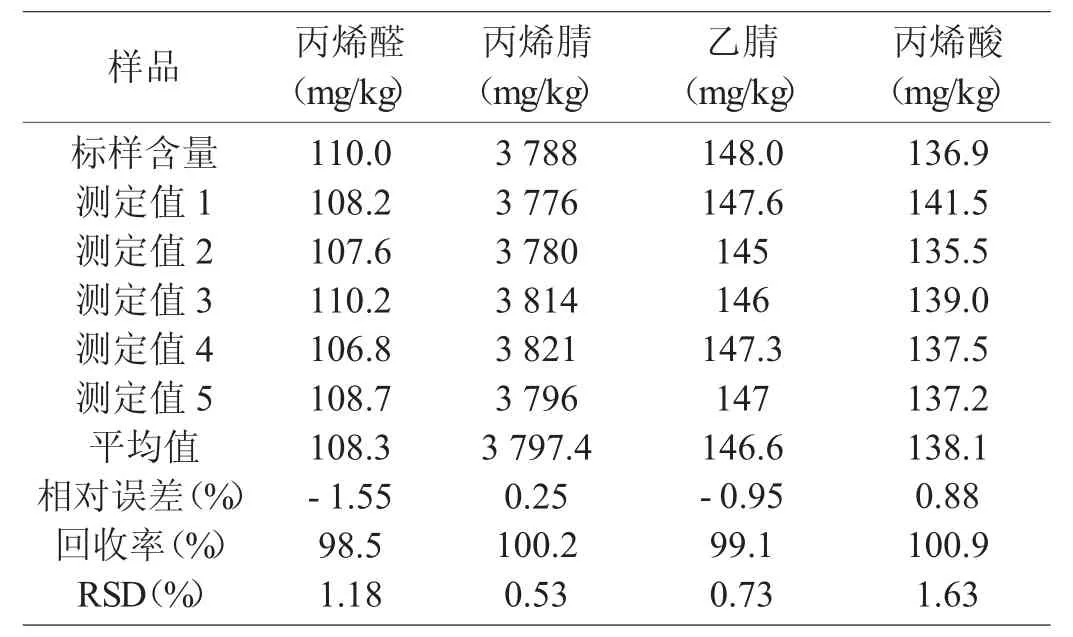
表4 标准样品准确度和精密度的验证
2.4 实际样品分析
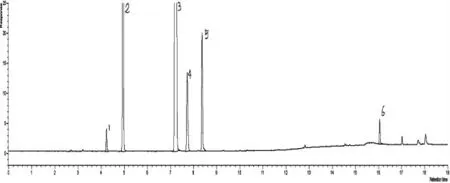
图4 DB-FFAP 毛细管柱分析丙烯腈反应器流出物实际样品色谱图
采用上述方法,用采集丙烯腈反应器吸收后的实际流出物样品,通过色谱分析,发现丙烯腈、乙腈、丙烯醛、丙烯酸分离效果较好,样品分析的重复性良好,样品典型色谱图见图4。
3 结论
(1)本方法改进了现有的丙烯腈反应器流出物测定方法,使用一台色谱代替两台色谱,毛细管色谱柱代替填充柱,操作简单,易掌握。
(2)采用DB-FFAP(60 m×0.32 mm×0.50 μm)毛细管色谱柱,通过程序升温实现一次分析成功。由于毛细管色谱柱分离效率高,缩短分析时间至20 min,提高了样品组分乙腈与丙烯醛的分离度,减少了丙烯酸的吸附。
(3)采用内标法定量,由两次进样改为一次,相对误差为-1.55%~+0.88%,回收率为98.5%~100.9%,准确度良好,相对标准偏差均小于2%,样品重复性好,使得丙烯腈单收数据准确可靠,为装置平稳运行和催化剂评价提供重要依据。
猜你喜欢
食品科学(2020年23期)2020-12-31 01:31:34
甘肃科技(2020年20期)2020-04-13 00:30:30
山东化工(2018年7期)2018-04-25 03:07:27
中国科技博览(2017年48期)2017-12-13 13:14:05
绿色科技(2017年16期)2017-09-22 20:11:13
海峡科技与产业(2017年3期)2017-04-13 14:16:07
石油化工技术与经济(2017年2期)2017-04-06 01:59:15
中国病理生理杂志(2017年2期)2017-01-17 03:59:14
化工生产与技术(2015年1期)2015-12-17 08:37:00
物理化学学报(2015年5期)2015-02-28 17:35:03
