
SPME-HPLC-MS同时检测尿液中氯胺酮与MDMA及其体内主要代谢物
2019-05-23 02:30:40李华云
中国药科大学学报 2019年2期
刘 红,李华云,龚 冶,孙 琴,4*,代 勇**
(1西南医科大学药学院;2西南医科大学附属医院药学部;3四川警察学院刑事科学技术系;4西南医科大学附属中医医院中西医结合药物研究中心,泸州 646000)
“摇头丸”(3,4-亚甲二氧基甲基苯丙胺为主要成分)和“K粉”(氯胺酮)是吸毒人员经常混合吸食的毒品,目前对混合毒品检测的报道主要集中在对原料药和缴获毒品的分析测定方面[1],对生物检材中毒品的检测大多针对单一毒品及其代谢物,其前处理通常采用液相萃取或固相萃取等方式[2]。近年来,固相微萃取作为一种快速便捷的提取方式,已广泛用于生物样品的前处理。在公安机关打击毒品犯罪的实际工作中,尿液因其易于采集、无损害且能反映毒品在体内代谢情况,已作为一种重要的生物物证,成为公安机关判断吸毒嫌疑人是否吸食毒品的首选检材[3]。目前,对混合毒品的检测大多采用气质联用技术进行检测,对实验条件有一定的要求。本研究采用固相微萃取-液相色谱-质谱联用技术,为此类混合毒品及其代谢产物的检测提供新的检测方法。
本研究以尿液中的氯胺酮与3,4-亚甲二氧基甲基苯丙胺(MDMA)及其主要代谢物去甲氯胺酮、3,4-亚甲二氧基苯丙胺(MDA)为检测对象,以固相微萃取作为样品前处理手段,并考察了其萃取条件,确定了HPLC-MS对尿液中氯胺酮、去甲氯胺酮、MDMA和MDA的检测方法,希望为公安机关提供较为便捷、准确的生物检材中混合毒品的鉴别方法,同时也为其他混合毒品及其体内代谢物的分析与检测提供参考与借鉴。
1 实验部分
1.1 仪器及测试条件
仪器:Agilent-1200 Series液相色谱仪,配G6130A质谱仪(美国Agilent公司);色谱柱为键合EC-C18柱(4.6 mm×100 mm,2.7 μm;美国Agilent公司)。萃取头:85 μm聚丙烯(PA)、100 μm聚二甲基硅氧烷(PDMS)、60 μm聚二甲基硅烷-二乙烯基苯共聚物萃取头(PDMS/DVB)、SPME进样器、CORNING固相微萃取采样台(美国Supelco公司);GPA224S万分之一分析天平(德国Sartorius公司);KH3200E型超声清洗仪(上海声源超声波仪器设备有限公司)。
色谱条件:采用EC-C18柱(4.6 mm×100 mm,2.7 μm)色谱柱,以0.1%甲酸水(A)-甲醇(B)作为流动相,梯度洗脱:0~6 min,70%A→30%A,6~7 min,30%A→10%A,保持3 min。流速0.2 mL/min,进样量1 μL,柱温40 ℃。
质谱条件:采用电喷雾电离源正离子扫描(ESI+),SIM监测模式;干燥气(N2)流速:10 L/min,干燥器温度:350 ℃,雾化电压3 kV,数据采集0~10 min,待检测物的SIM监测离子和碰撞能见表1。
Table1 SIM of analyte monitor ions and collision energy
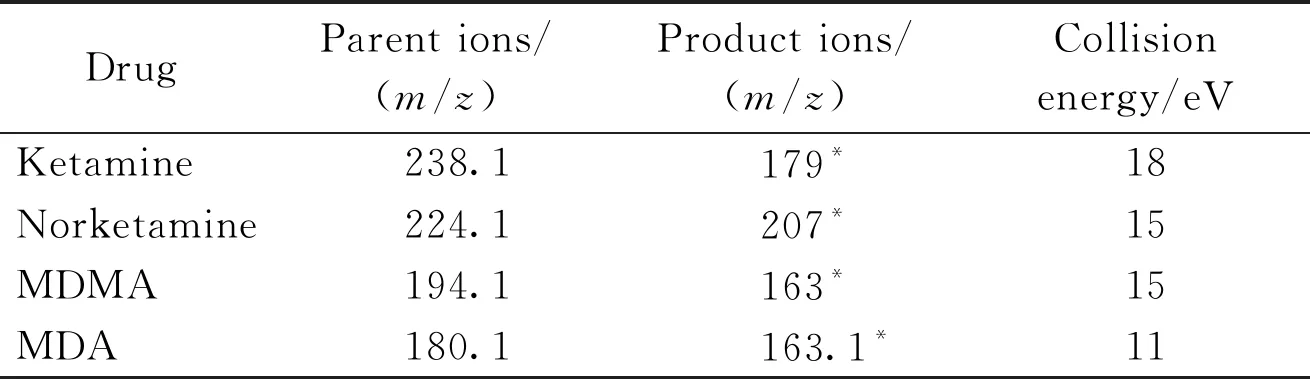
DrugParent ions/(m/z)Product ions/(m/z)Collision energy/eVKetamine238.1179*18Norketamine224.1207*15MDMA194.1163*15MDA180.1163.1*11
*Quota ion
MDMA:3,4-Methylenedioxymethamphetamine;MDA:3,4-Methylenedioxyamphetamine
1.2 药品与试剂
对照品氯胺酮、去甲氯胺酮、MDMA、MDA(均为甲醇液,质量浓度为1.00 mg/mL)均由泸州市公安局提供。甲醇、乙腈均为色谱纯,盐酸、碳酸钠和氢氧化钠等其余试剂均为分析纯,水为纯化水。
对照储备溶液的制备:分别取氯胺酮、去甲氯胺酮、MDMA、MDA标准品甲醇液1 mL,稀释为100 mL,置于4 ℃冰箱保存。
1.3 实验动物
清洁级别大鼠,重量(200±10)g,雌雄各半,西南医科大学动物实验中心提供(使用许可证号:SCXK(川)2018-17)。给药前禁食不禁水12 h,灌胃给药。
1.4 尿液样品的制备
取健康人空白尿液10 mL,分别添加氯胺酮、去甲氯胺酮、MDMA和MDA对照储备液,配制成氯胺酮、去甲氯胺酮、MDMA、MDA均为1 μg/mL的毒品尿液样品,备用。
1.5 操作方法
取尿液样品5 mL于10 mL样品瓶中,用1.25 mol/mL HCl和NaOH溶液调节pH至11,加入Na2CO30.5 g,放入磁搅拌子,拧紧瓶盖,将样品瓶置于SPME专用采样台上于60 ℃下加热搅拌,同时将经老化过的含60 μm聚二甲基硅烷-二乙烯基苯共聚物萃取头(PDMS/DVB)的SPME针管插入样品瓶中,推出萃取纤维头,使之浸入样品液中萃取15 min后取出,再将SPME针管插入SPME/HPLC接口解吸池中解析1 min后,进行数据采集。
2 结果与讨论
2.1 色谱柱及流动相的考察
预实验中比较了C18柱(2.1 mm×50 mm,1.8 μm)和C18柱(4.6 mm×100 mm,2.7 μm)的检测效果,发现因所检测的氯胺酮和MDMA等物质含有胺基和羰基等基团,为中等偏强极性物质,所以其保留时间较短,均在0.6 min左右出峰,为了提高分离效果,最终选择了EC-C18柱(4.6 mm×100 mm,2.7 μm)作为分析柱。
氯胺酮、去甲氯胺酮、MDMA和MDA的保留时间、分离度和峰行受流动相体系及其PH的影响。本实验分别考察了甲醇/乙腈-水、甲醇/乙腈-甲酸水溶液、甲酸乙腈-水溶液和甲酸乙腈-甲酸溶液体系等作为流动相。结果表明,在酸性条件下,有助于化合物母离子的形成。比较甲酸体积分数为0.1%、0.2%、0.3%时,发现增大甲酸浓度对化合物检测灵敏度影响不大,因此,最终选择甲醇和0.1%甲酸水溶液体系作为流动相。
按以上色谱柱与流动相对混合毒品进行检测,氯胺酮、去甲氯胺酮、MDMA和MDA的保留时间分别为6.52、6.60、4.99、5.03 min,见图1。
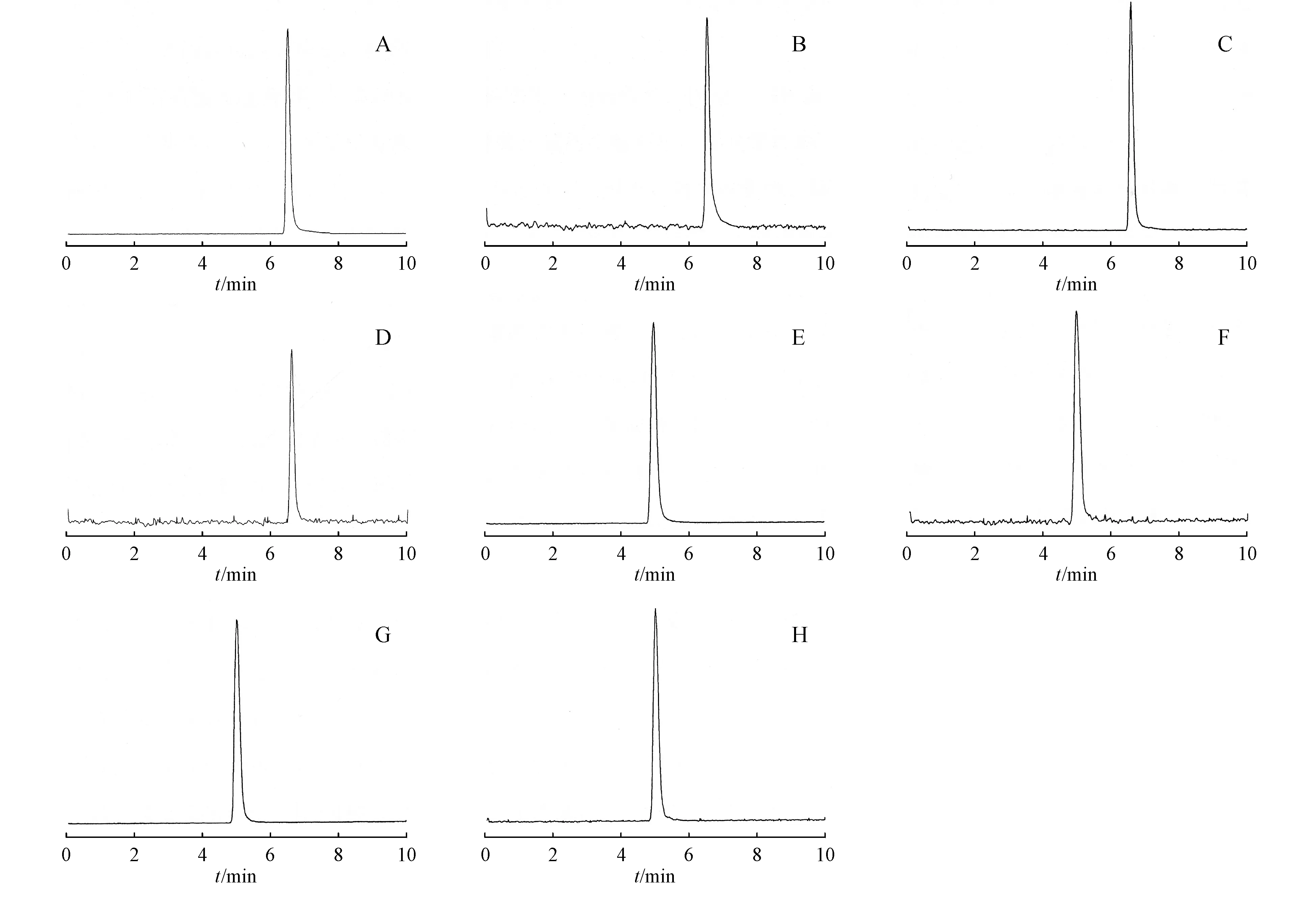
Figure1 SIM chromatogram of ketamine,norketamine,MDMA,and MDA
A:Ketamine,m/z238.1;B:Ketamine,m/z179;C:Norketamine,m/z224.1;D:Norketamine,m/z207;E:MDMA,m/z194.1;F:MDMA,m/z163;G:MDA,m/z180.1;H:MDA,m/z163.1
2.2 固相微萃取前处理影响因素考察
2.2.1 萃取纤维种类和萃取模式考察 在实验中主要选择了3种不同极性且适合于HPLC进样分析的萃取头进行考察:85 μm聚丙烯(PA) 、100 μm聚二甲基硅氧烷(PDMS)、60 μm聚二甲基硅烷-二乙烯基苯共聚物萃取头(PDMS/DVB),在相同的萃取条件下进行直接浸入式(DI-SPME)萃取,以峰面积大小作为萃取效果的评价指标,结果表明,由于PA萃取头对强极性的物质富集效果更好,PDMS萃取头对弱极性物质的萃取效果更好,故这两种萃取头的萃取效率略低。比较而言,PDMS/DVB共聚物极性介于前两者之间,且适合于对胺类化合物的提取,故其萃取效率较前两者高。
固相微萃取分为直接浸入模式(DI-SPME)和顶空模式(HS-SPME)两种方式,由于本次检测目的是以HPLC为检测手段对两种毒品在体内的微量原型物和代谢物样品进行分析,故选用直接浸入模式,提高萃取效率。
2.2.2 萃取溶液酸碱度考察 本次检测的4种物质均为弱碱性,因此在碱性条件下,有利于减少其在基质中的电离度和溶解度,促进其从基质中解析出,提高萃取率。为此,本研究考察了pH为9,10,11,12,13的不同条件下,对萃取效果的影响。随着pH的增加,各物质的萃取率均存在不同程度的上升,在pH为11以后时,4种物质的萃取率基本保持恒定,且尿液基质中开始出现少许沉淀(可能在较强碱性条件下,其中的蛋白质成分变性所致),故选择萃取酸碱度为pH 11。
2.2.3 萃取温度和时间考察 查阅相关文献[4],发现温度对萃取效果有较大的影响,其机制是通过改变目标成分在溶液与萃取介质间的分配系数,从而影响萃取纤维对目标成分的吸附效率。随着温度升高,溶液中目标成分的热运动增加,目标成分的分子向萃取纤维扩散的概率大大提高,从而达到富集目标成分的目的。但是,当温度过高时,将导致萃取纤维中的目标成分向溶液中扩散,破坏了分配平衡,这种反相扩散有可能导致萃取的效率锐减。因此,在实验过程中,须筛选适宜的温度,才能取得较好的萃取效果。
在30~70 ℃区间范围内,以10 ℃递增进行萃取温度条件考察,其结果如图2所示,4种目标物质的萃取效率随着温度改变呈现先上升后下降的现象,因各物质结构间存在差异,其最佳萃取温度并不一致,综合考虑具体情况,选择萃取温度为60 ℃,同时,在萃取时加入搅拌子以增加待测物质在基质溶液中的热运动,改善萃取平衡系数。


目标物质在基质溶液与萃取介质间的平衡时间受其物理化学性质(如化学结构、在两相间的分配系数等)、基质条件和萃取介质吸附特性等多种因素决定,萃取时间短,介质吸附不充分,未达到分配平衡,目标物回收率较低,萃取时间过长,实验费时,达不到优化实验条件的目的。
在5~25 min区间内,以每5分钟为一检测点,峰面积为检测指标,选择合适的萃取时间,其结果如图3所示。由于4种目标物质相对分子质量较小,在基质与萃取介质间的能较快地达到平衡,故选择萃取时间为15 min。

2.3 方法学考察
2.3.1 线性范围及检测限 取空白尿液检材5份,添加氯胺酮、去甲氯胺酮、MDMA、MDA对照品,用流动相稀释,配制成0.03、0.20、0.50、0.75和1.0 μg/mL系列质量浓度对照品的尿液样本,按以上所述实验方法进行目标物提取与检测分析,以待测物定量离子峰面积Y为纵坐标,以所配制的系列对照品浓度X为横坐标,汇制标准曲线,得到4种待测物的线性方程、其各自的相关系数r和最低检出限(检测时信噪比为S/N=3)见表2。结果表明,4种待测物在0.03~1.0 μg/mL范围内线性关系良好。
Table2 Linear equations,correlation coefficient and detection limit (S/N,n=3)

AnalyteLinear regression equationrDetection limit/(ng/mL)KetamineY=5.527×104X-4.150×10-10.999 211.2NorketamineY=7.769×104X-8.998×10-10.999 110.8MDMAY=3.000×105X-6.103×10-20.998 913.6MDAY=3.850×105X-1.266×10-20.998 514.3
2.3.2 加样回收试验及精密度考察 取空白尿液,分别加入高、中、低3个浓度的对照品,配制成5个平行样品,按照“1.1”、“1.5”项下进行检测分析,连续测定3 d,并记录色谱峰面积,计算氯胺酮、去甲氯胺酮、MDMA、MDA的日内、日间精密度,结果如下表3所示。
在4种待测物线性方程范围内,取高、中、低3个浓度分别添加对照品,配制成5个平行样本检材,然后按“1.1”、“1.5”项下进行样品处理、分析。以所添加待测物的峰面积与相同浓度标准品的峰面积之比值计算回收率(结果见表3)。
Table3 Results of recovery and reproducibility test (n=5)
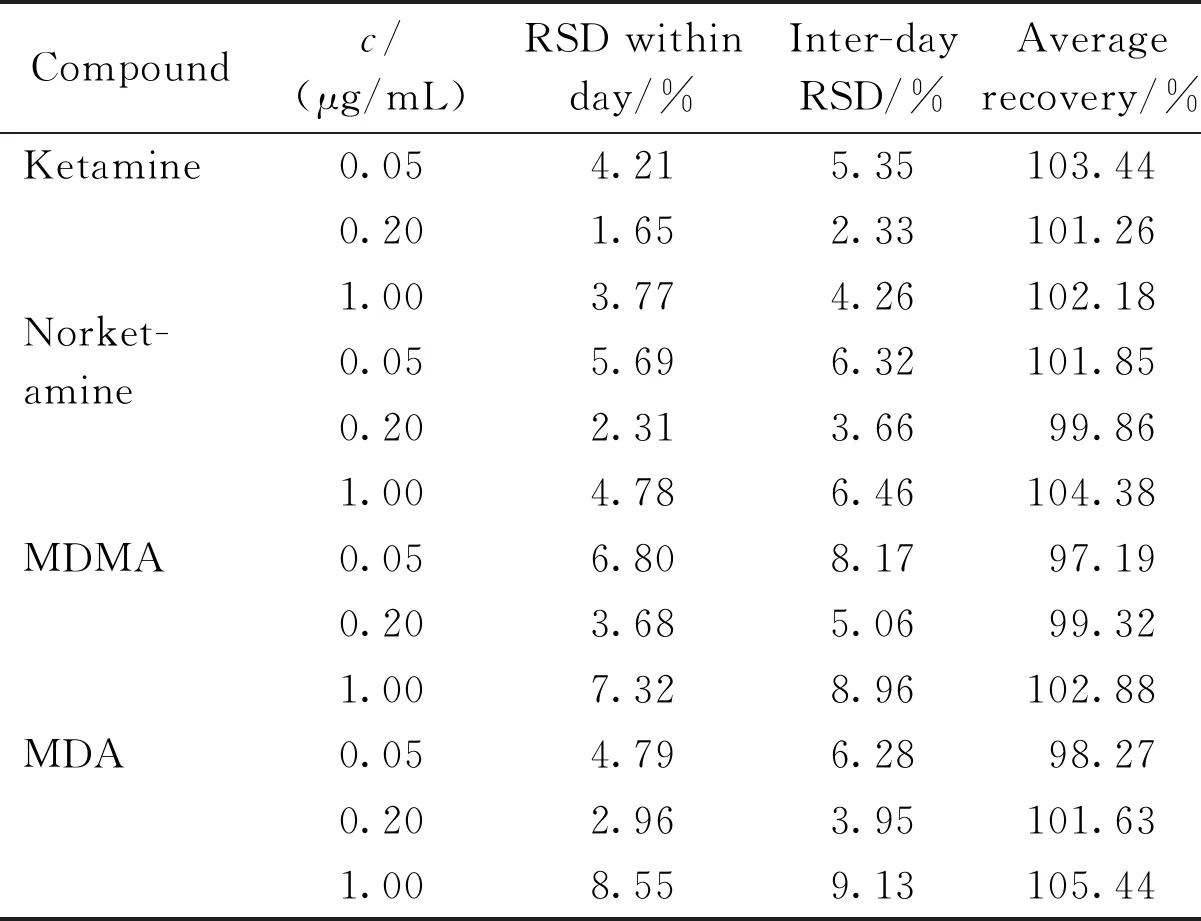
Compoundc/(μg/mL)RSD within day/%Inter-day RSD/%Average recovery/%Ketamine0.054.215.35103.440.201.652.33101.261.003.774.26102.18Norket-amine0.055.696.32101.850.202.313.6699.861.004.786.46104.38MDMA0.056.808.1797.190.203.685.0699.321.007.328.96102.88MDA0.054.796.2898.270.202.963.95101.631.008.559.13105.44
2.4 样品分析
据文献文献[5-6]报道,大鼠以氯胺酮290 mg/kg灌胃给药后,出现急性中毒现象,大鼠灌胃给药150 mg/kg MDMA后,也同样出现急性中毒。根据现在毒品的吸食模式,综合考虑动物生命周期,取喂食“K粉”与“摇头丸”粉末(“摇头丸”研磨成粉状)(氯胺酮按280 mg/kg灌胃,MDMA按140 mg/kg灌胃)的急性中毒大鼠24 h内自然排泄尿液样品5 mL,按以上所述试验方法进行检验,检材中分别出现与对照品相对应的质谱图,因此可认定检材中存在氯胺酮、去甲氯胺酮、MDMA和MDA 4种成分。进一步测定得出该尿液样本中的氯胺酮、去甲氯胺酮、MDMA和MDA的含量分别为2.02、3.01、1.05、1.98 μg/mL。
3 结 论
本试验采用SPME-HPLC-MS法对尿液中的“K粉”与“摇头丸”混合毒品及其体内主要代谢产物进行了定性和定量分析,本方法操作便捷,结果准确,可为公安机关对吸毒嫌疑人的尿液等生物样品进行检测提供新的方法,为公安机关打击毒品犯罪提供法庭证据。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13 14:57:02
中国神经精神疾病杂志(2020年9期)2020-01-14 19:02:12
现代养生·下半月(2017年10期)2017-03-23 16:28:28
西南军医(2016年3期)2016-01-23 02:17:58
合成化学(2015年10期)2016-01-17 08:56:07
中国继续医学教育(2015年6期)2016-01-07 07:38:47
分子影像学杂志(2015年3期)2015-12-04 03:28:54
海峡科学(2015年11期)2015-09-19 06:48:18
中国民族民间医药·下半月(2014年2期)2014-09-26 05:21:22
同位素(2014年2期)2014-04-16 04:57:16
