
基因表达数据特征子集的冗余研究
2019-05-11 07:49周长银
统计与信息论坛 2019年5期
杜 冲,周长银
(山东科技大学 数学与系统科学学院,山东 青岛 266590)
一、引言
随着科学技术的快速发展,我们能够获得越来越多的微阵列数据。依据这些数据进行正常组织与癌症组织的诊断,或者不同癌症组织的判别分析已经在生物学领域得到了广泛的应用。基因表达数据具有小样本、高维度、分布不平衡的特点,因此,如何有效地处理、利用与分析这些数据,成为我们面临的一个关键问题,而且大多数基因并不能为疾病诊断提供有用的信息,这些信息只包含在少数的基因之中。例如,Golub等人在对急性淋巴细胞白血病(ALL)和急性髓性白血病(AML)两类白血病进行研究时,发现50个特征基因能够包含足够的信息,并能对所有信息进行分类[1]。Alon等人对结肠癌数据集进行研究时,选择了20个最具有统计学显著差异的特征基因,并取得了良好的分类效果[2]。因此,对基因表达数据进行特征选择具有很多优点:一方面可以降低数据维度减少计算量,另一方面可以减少噪声的影响,提高分类准确度。
特征选择已经成为生物信息学领域数据预处理步骤不可或缺的一部分,特征选择技术可以宽泛地分为过滤式(Filter)方法、封装式(Wrapper)方法、嵌入式(Embedded)方法三类[3]。过滤式方法只通过数据的内在属性来估计特征的差异性,而不考虑模型的学习算法或分类器对特征的影响。过滤式方法的常用方式是根据特征的差异性评分进行排序,并选取评分较高的一部分特征作为特征子集输入到分类算法上。一些常用的过滤式特征选择方法如信息熵、t检验、χ2检验与秩和检验都显示了良好的性能[4-7]。封装式方法通过评估分类器的分类性能在候选子集空间内选取最佳的特征子集,对于特定的学习算法,封装式方法可能会取得比过滤法更好的效果,但会增加计算成本。嵌入式方法将特征选择方法嵌入到过滤器中,通常能够在计算成本和分类性能之间达到一个很好的平衡。过滤式方法计算简单快速,独立于分类算法,因此对于高维度数据集能够极大地减少运算成本,并且适用于不同的分类算法。由于技术成本问题,基因表达数据集的样本数量往往在100左右,但基因的数量即特征的数量高达成千上万个,原始数据集经过过滤式特征选择后获得的特征子集仍然具有较高的维度。过滤式特征选择方法没有考虑不同特征之间的相关性,因此,如果一个特征被选入特征子集,那么与此特征高度相关的一些特征也可能被选入特征子集,这些高度相关特征提供的分类信息往往是相似的,这会造成特征子集的冗余。这种冗余不仅不能为分类模型提供有用的信息,而且会成为一种噪声影响分类模型的准确度。对于这一问题,本文提出一种启发式的冗余去除算法。
此算法首先使用过滤式特征选择方法对原始基因表达数据集进行特征选择,获得特征子集,然后在特征子集的基础上开展冗余去除的工作。在特征子集冗余去除之前,要确定不同变量之间的相关性度量,本文选取了皮尔逊相关系数。由于不同强度相关系数会对冗余去除与分类效果形成不同影响,因此具体分析了这一因素对整体模型的影响。为了验证算法的有效性,获得科学合理的实验结果,本文在白血病、结肠癌和前列腺疾病三个基因表达数据集上,使用支持向量机(SVM)、k近邻、随机森林三种不同的分类器分别进行了测试[8-10]。
二、过滤式特征选择的统计方法
对于二分类问题,一种常用的过滤式特征选择方法是使用统计学中的假设检验。假设检验可以分为参数检验和非参数检验,参数检验首先需要确定总体的分布类型,然后针对参数进行假设检验,常用的方法有t检验、F检验和χ2检验,非参数检验不需要假定总体的分布类型,直接对总体分布的某种假设(例如对称性、分位数大小)做统计检验,常用的方法有秩统计量、符号秩统计量[11]。给定一个基因表达数据集G(包含m+n个样本,N个基因),其中X={x1,x2,…,xm}为正类样本,Y={xm+1,xm+2,…,xm+n}为负类样本,特征基因集合为G={g1,g2,…,gN},gij为第i个样本第j个特征的基因表达水平数值。设定显著性水平α,使用假设检验方法对数据集中每一个基因判断在此显著水平下是否通过假设检验,如果拒绝原假设,说明此基因在不同类别的总体分布存在差异,可以为分类模型提供有效的信息。将这些基因加入特征子集,作为分类模型的输入变量。本文在实验验证过程中分别使用了参数检验中的t检验方法和非参数检验中的秩和检验方法。
(一)t检验
t检验是适用于检验正态分布样本平均值差异的一种方法,它是用t分布理论来推断差异发生的概率,从而判定两个变量平均数的差异是否显著。对于基因表达数据特征选择问题,t检验首先需要建立原假设H0∶μ(gXj)=u(gYj),即假定两类样本在第j个特征gj上的总体平均数之间没有差异。为了评价两组样本平均数之间的差异程度,计算t统计量的值:
(1)
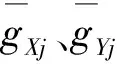
(二)秩和检验
Wilcoxon秩和检验是用来检验两个独立样本是否来自相同或相等的总体。Wilcoxon秩和检验基于样本数据的秩和,先将两样本看成单一样本(混合样本),然后由样本值从小到大排序统一编秩。如果原假设两个独立样本来自相同的总体为真,那么秩将大约均匀分布在两个样本中,因此获得的秩和统计量不会过大或过小,设符号函数:
i=1,2,…,m;
k=m+1,m+2,…,m+n
(2)
则Wilcoxon秩和统计量定义为:
(3)
但是过滤式特征选择方法存在一个较大的缺点,即在特征选择过程中没有考虑不同特征之间的相关性。例如,如果基因gi拒绝了原假设,被选入特征子集,那么与gi高度相关的一些基因也有可能被选入到特征子集中。这样获得的特征子集具有大量的冗余,当构建分类器时,这些冗余会成为噪声影响特征子集的分类性能,因此,如何去除特征子集的冗余是需要解决的一个重要问题。
三、特征子集的冗余去除模型
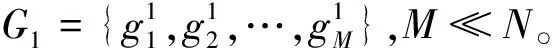
(一) 皮尔逊相关系数
在统计学中,皮尔逊相关系数用于度量两个变量之间的线性相关程度,其值介于-1与1之间[12]。对于特征基因gi与gj,样本相关系数ρij可以通过以下公式计算:
(4)
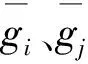
(二)冗余去除模型
我们希望去除特征子集中的冗余特征,使特征子集中不同的特征能够提供不同的分类信息,从统计角度看,这要求不同特征之间不能有太高的相关性,即对于某一特征来说,它和特征子集中其他的特征之间应该都不具有较强的相关性,因此,本文使用两两配对的方式依据皮尔逊相关系数去除特征子集中的冗余特征。
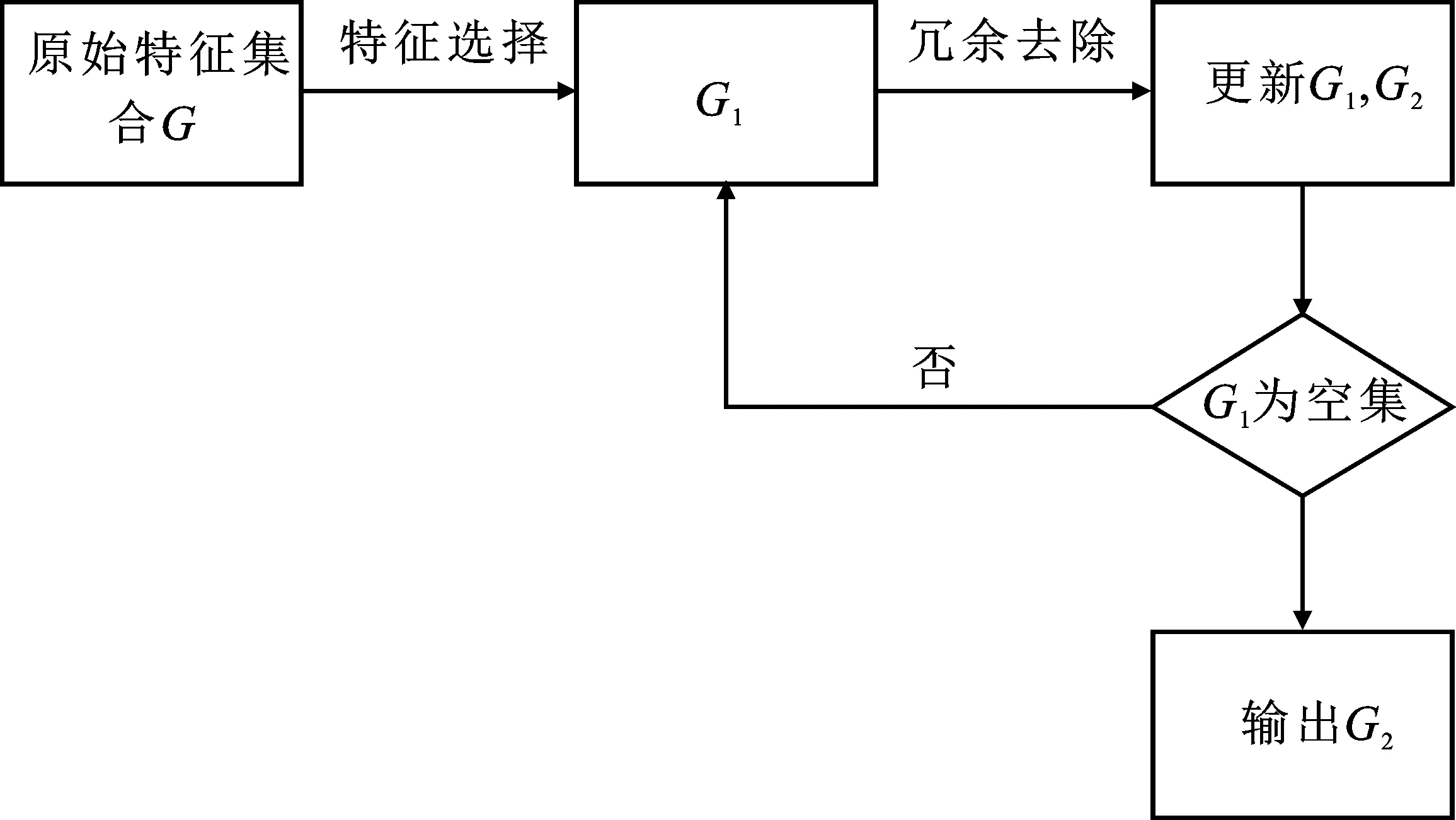
图1 冗余去除流程图
四、带冗余去除的特征基因选择算法
本文基于基因表达数据,首先使用统计学中假设检验的过滤式特征选择方法对数据集进行特征选择,然后对特征选择后的特征子集进行冗余去除。因此,带冗余去除的特征基因选择算法分为两个步骤:特征选择与冗余去除。
(一)特征选择算法
输入:基因表达数据集G,假设检验p值的阈值P。
输出:特征子集G1。
1.设置G1为空集;
2.对特征基因gi,i=1,2,…,N进行假设检验,计算假设检验p值pi;
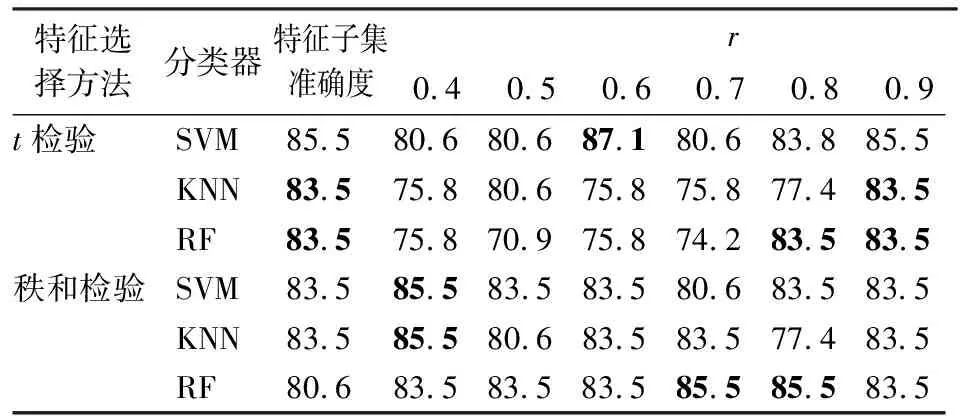
3.如果pi 4.输出特征子集G1。 输入:基因表达数据集G,特征子集G1,皮尔逊相关系数阈值r。 输出:冗余去除后的特征子集G2。 1.设G2为空集; 2.计算特征子集G1的长度l; 3.计算特征子集G1中第一个特征与其他特征的相关系数绝对值ρ1i,2≤i≤l; 4.如果ρ1i≥r则将第i个特征从特征子集G1中去除,否则特征子集G1保持不变; 5.去除特征子集G1中的第一个特征并保存在G2中,更新G1; 6.如果G1为空集,则输出G2,否则转入步骤3。 本文使用白血病、结肠癌和前列腺疾病三个基因表达数据集,数据集具体信息如表1所示。Colon数据集为结肠癌数据集,正类为结肠癌样本,负类为正常组织样本。Leukemia数据集为白血病数据集,正类为急性淋巴细胞白血病(ALL)样本,负类为急性髓性白血病(AML)样本。Prostate数据集为前列腺疾病数据集,正类为前列腺样本,负类为正常组织样本。“来源”中的序号为文后参考文献序号。为了评价特征子集的分类预测性能,使用支持向量机(SVM)、k近邻、随机森林三种分类器分别进行测试。 表1 基因表达数据集 在实验过程中,使用统计学假设检验方法进行特征选择时假设检验阈值P设定为0.05。一般认为皮尔逊相关系数在[0,0.4]之间变量为弱相关,[0.4,0.7]之间为中等相关,[0.7,1.0]之间为强相关,因此本文将皮尔逊相关系数阈值r设定在[0.4,0.9]。支持向量机的核函数选用线性核,考虑样本数量k近邻中k值在Colon与Leukemia数据集中设置为3,在Prostate数据集中设置为5,随机森林中树的数量设定为100。为预防过拟合现象和人为因素的影响,对数据集进行五折交叉验证实验。 模型算法使用R软件 x64 3.3.3版本的脚本语言编写,并在Windows 7上运行。 在进行特征子集的冗余性分析之前,需要对基因表达数据集进行特征选择,图2显示了不同数据集使用t检验与秩和检验的特征选择结果,结果均为交叉验证实验结果的均值。 Colon数据集原始特征数目为2 000,经过t检验特征数目降低到415.6,经过秩和检验特征数目降低到325.6;Leukemia数据集原始特征数目为7 129,经过t检验特征数目降低到1 815.8,经过秩和检验特征数目降低到1 855.4;Prostate数据集原始特征数目为12 600,经过t检验特征数目降低到4 448,经过秩和检验特征数目降低到3 863。可以看出,t检验与秩和检验在特征选择的降维效果上差别不大,经过特征选择后特征集合的数目大为减少,但特征子集的数目还是比较庞大。在特征子集中存在大量彼此相关的特征,因此需要对特征子集的冗余性作进一步研究。 图2 t检验与秩和检验特征选择结果对比 图3中横坐标t与w分别代表t检验与秩和检验,数字代表设定的相关系数阈值r。为方便比较,r取值从大到小排列。 图3 冗余去除特征数目对比 由图3可知,不同数据集在同一阈值下会得到不同的冗余去除效果。整体来看,经过t检验与经过秩和检验获得的特征子集在冗余去除上具有相同的表现趋势。Colon数据集与Prostate数据集在r=0.8时会去除大量冗余得到一个较小的特征子集,说明在这两个数据集的特征子集中多数特征之间具有强相关性,稍有不同的是Prostate数据集表现了更强的相关性,一大部分特征之间的相关性大于0.9。Leukemia 数据集在r=0.6时去除大量冗余得到一个较小的特征子集,并且在r=0.9与r=0.8时冗余去除效果不明显,说明Leukemia数据集的特征子集中多数特征之间没有强相关性,但具有中度相关。 为了检验特征选择后的特征子集及冗余去除后的特征集合的分类预测性能,使用支持向量机(SVM)、k近邻(KNN)、随机森林(RF)三种分类器分别进行了测试,分类准确度结果如表2~4所示,加黑部分为同一分类器下达到的最高准确度。可以看出对于不同的基因表达数据集,分类的最高准确度大多数情况下是在去除冗余后的特征子集中获得的。这说明经过过滤式特征选择后的特征子集中存在大量冗余,这些冗余不仅会增加特征子集的维度,还会降低分类器的准确度。另外在相关性阈值r取值为0.4或0.5时,分类准确度相对于最高值虽然会有所降低,但也可以达到一个较好的水平,而且此时冗余去除后的特征子集的维度会降低到一个极小的水平。这表明,在成千上万个基因中只有极少数关键性的基因对疾病分类起到决定性作用。 表2 Colon数据集分类准确度 表3 Leukemia数据集分类准确度 表4 Prostate数据集分类准确度 特征选择是基因表达数据预处理过程中关键性的步骤。本文对过滤式特征选择后特征子集的冗余做了进一步研究,将特征子集冗余去除模型加入到特征选择中,构建带冗余去除的过滤式特征选择模型。此模型在解决冗余性问题的同时降低了特征子集的维度,有助于减少后续分类模型的计算量,节约了计算成本和存储成本。在对实际数据集进行验证时,本文模型能够达到一个较为理想的实验效果,验证了特征子集冗余去除的必要性,并且在实验过程中发现,分类器在极少数特征下就可以达到一个较好的分类准确度,这也符合生物学的基本原理,疾病的产生只和少数基因有关。因此,特征子集冗余去除工作对于基因表达数据集具有重要的意义。另外,如何更加精准地定位这些极少数的基因,并在此基础上获得更加准确的疾病诊断,是下一步研究的方向。 RedundantStudyonFeatureSubsetofGeneExpressionData DU Chong,ZHOU Chang-yin (School of Mathematics and Systems Science,Shandong University of Science and Technology,Qingdao 266590,China) Abstract:Filter feature selection is a widely used method on gene expression data which is also simple and effective.Aiming at the redundancy of its feature subset,a feature selection algorithm with redundancy elimination is proposed by using Pearson correlation coefficient.Through the algorithm,the redundancy removal and classification accuracy of feature subset under different correlation intensities are studied.Three different gene expression data sets are selected and tested using support vector machine,nearest neighbor and random forest as classifier.The experimental results show that the filtering feature selection method with redundant removal can achieve good classification performance on different classifiers.In addition,using this method can improve the classification accuracy while reducing the feature subset dimension. Keywords:gene expression data; feature selection; filtering method; Pearson correlation coefficient; redundancy removal(二)冗余去除算法
五、实验及结果
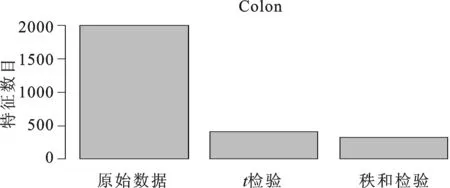
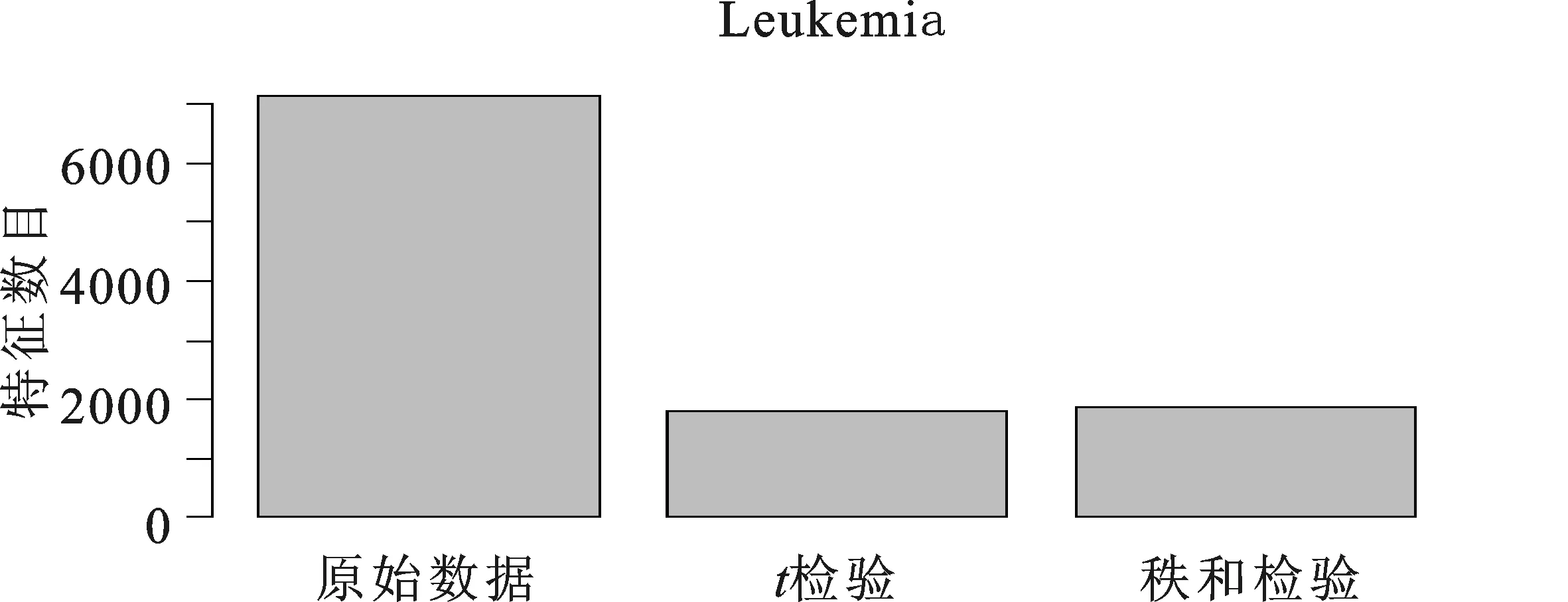
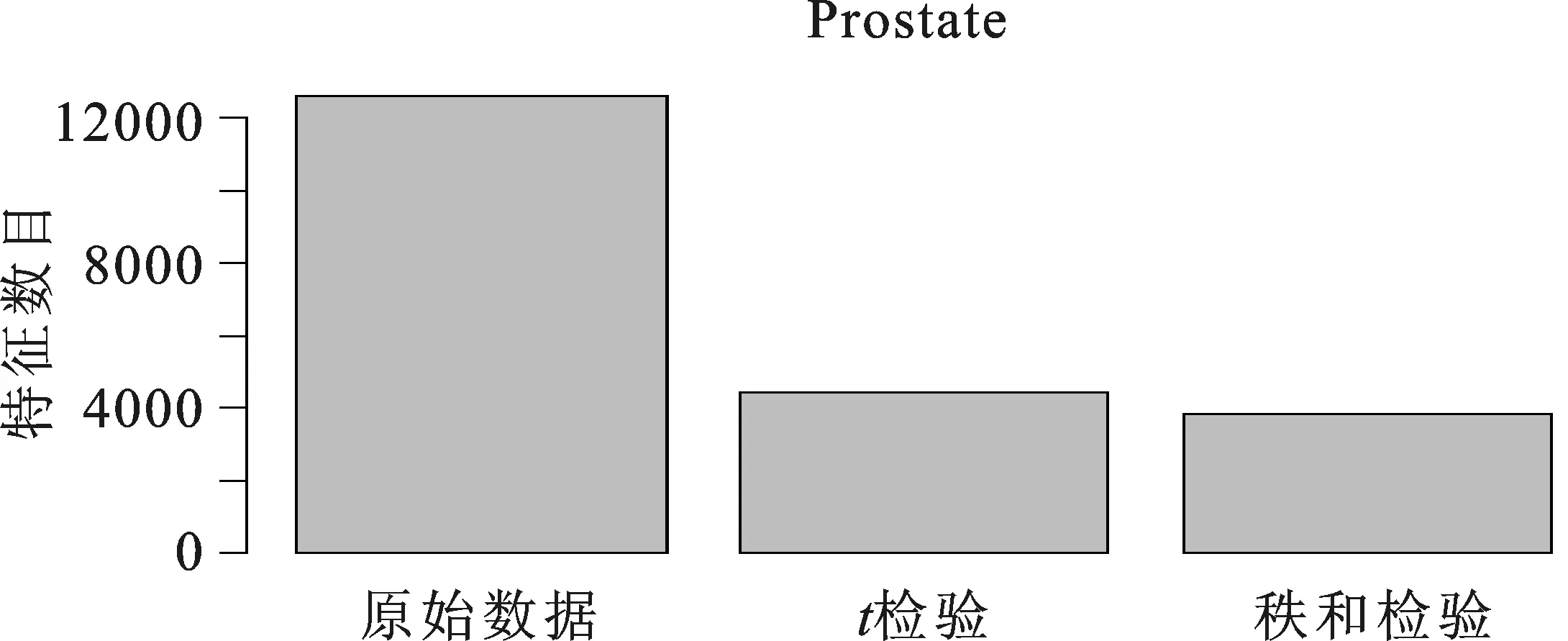
(一)特征选择结果
(二)冗余去除效果
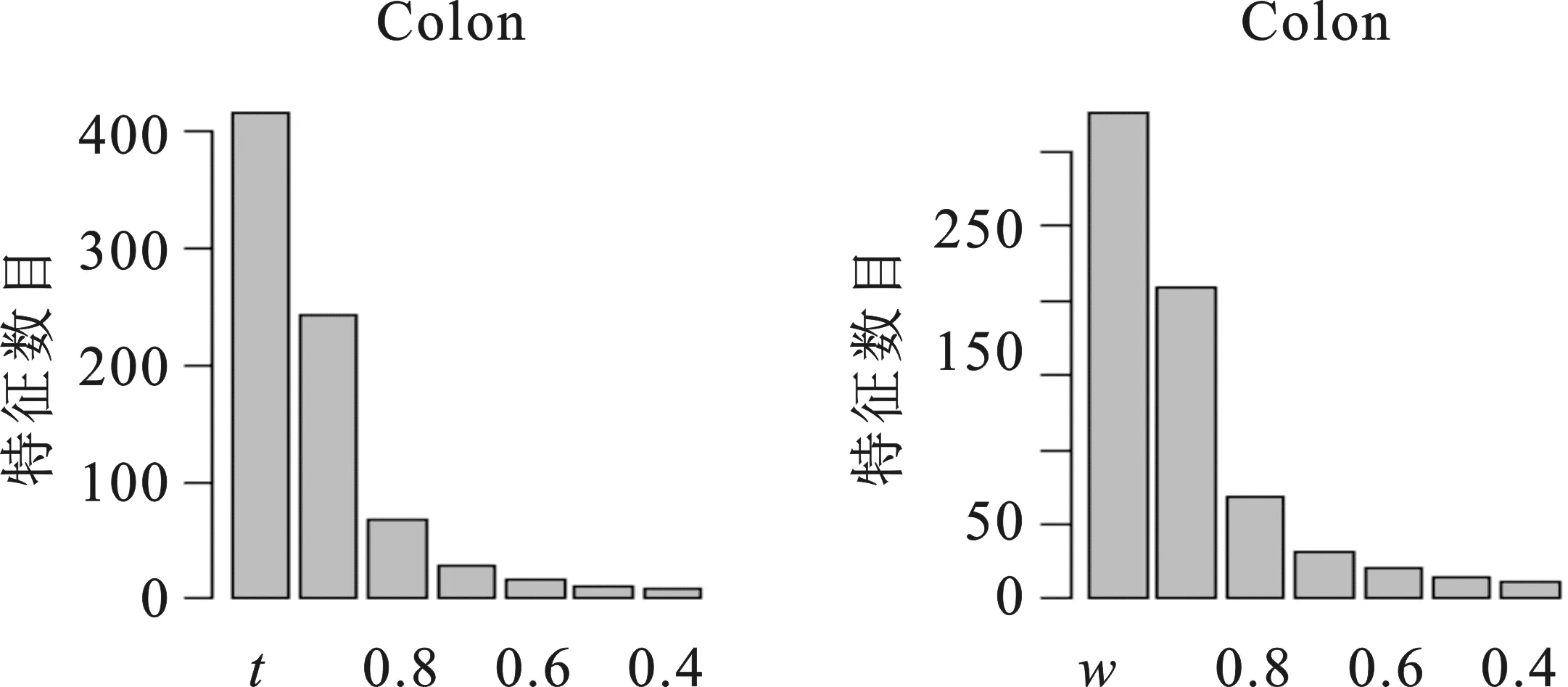
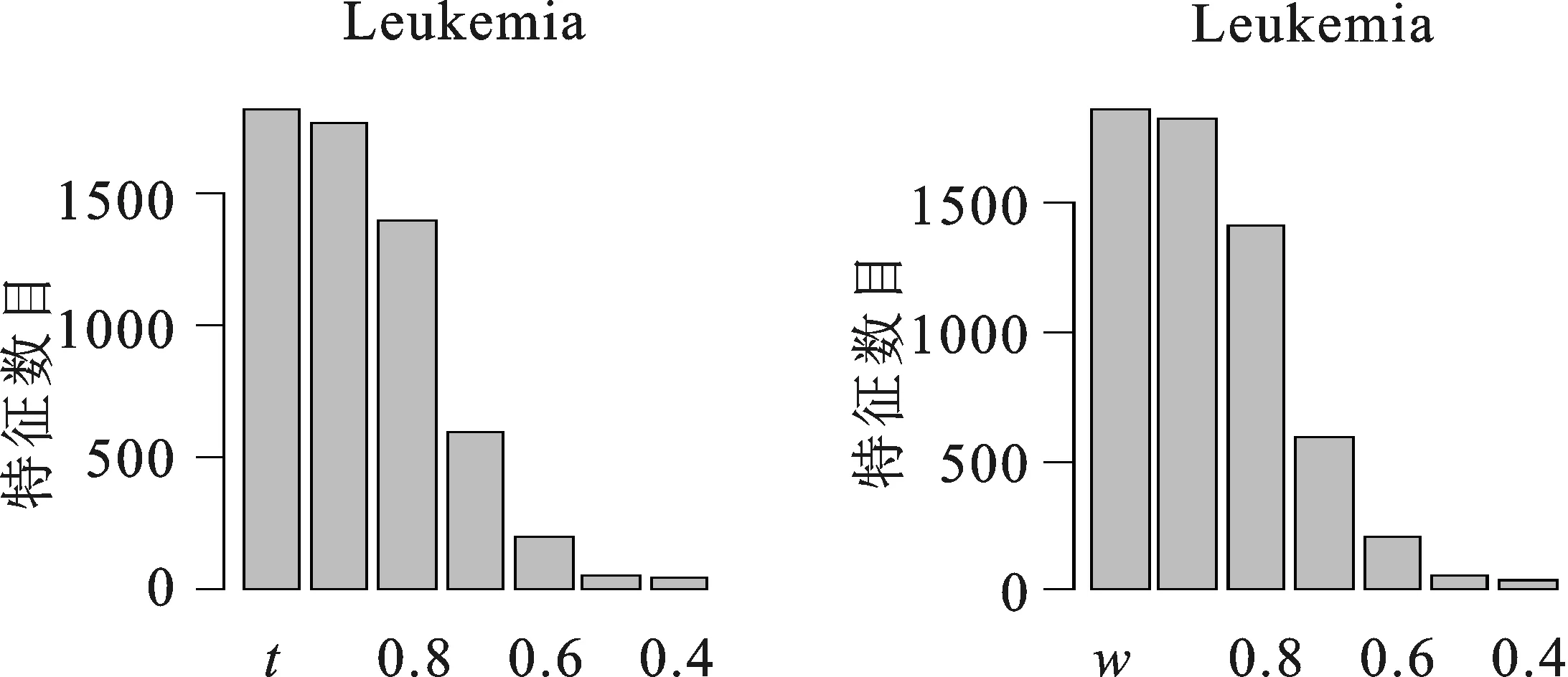
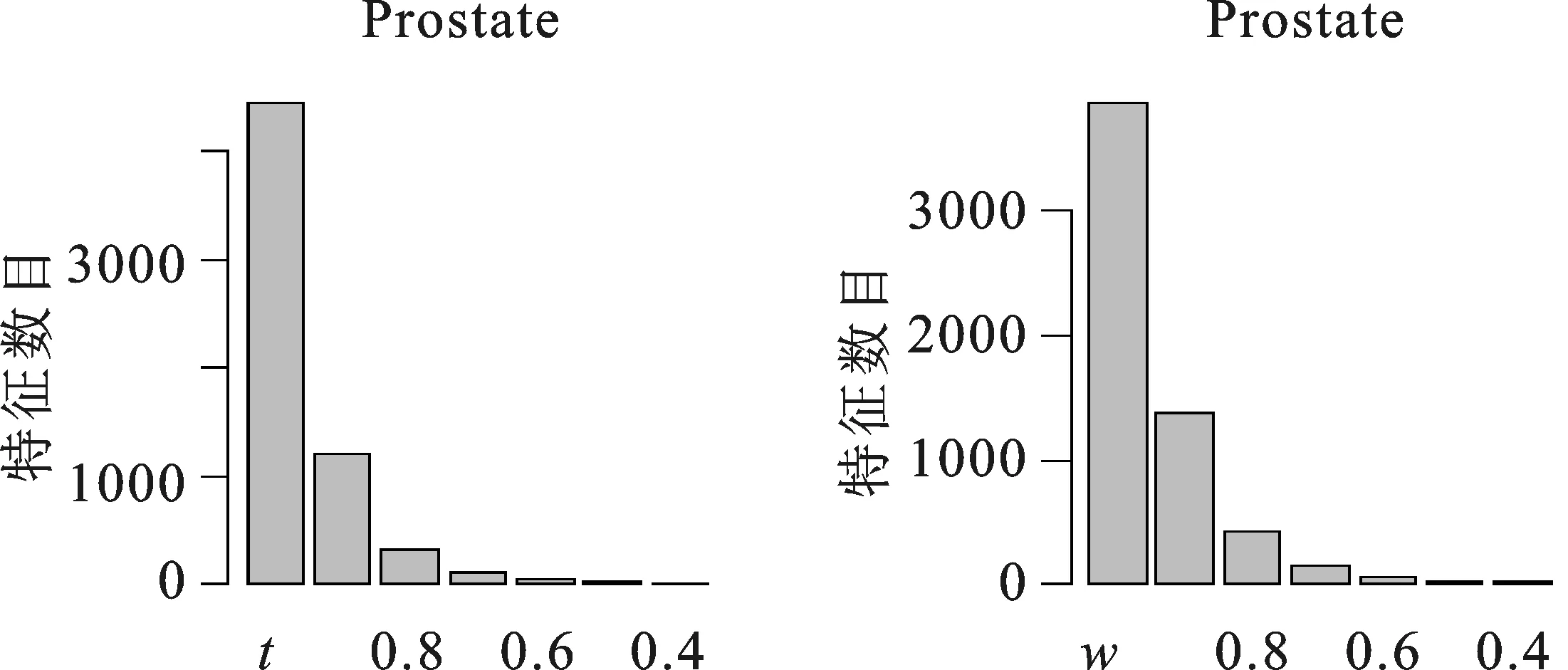
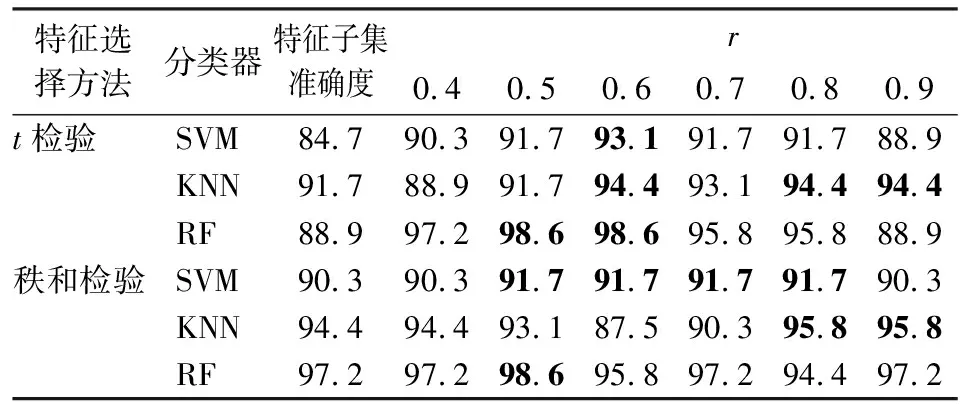
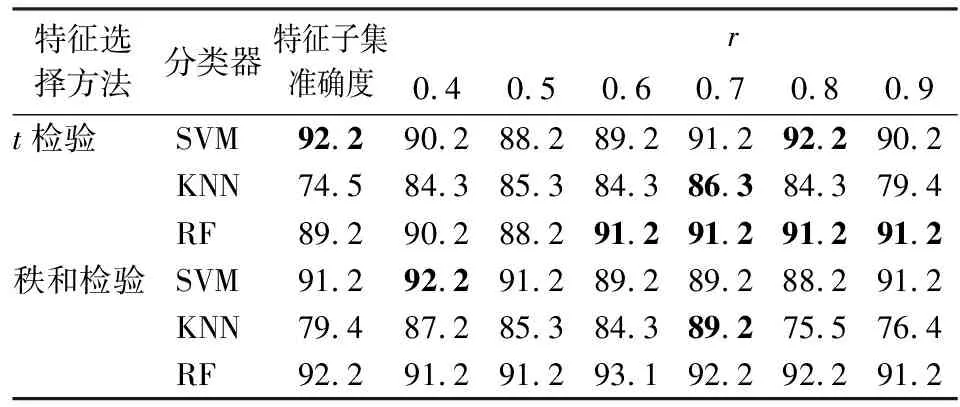
(三)分类准确度
六、结语
猜你喜欢
福州大学学报(自然科学版)(2022年1期)2022-01-21
安庆师范大学学报(自然科学版)(2021年1期)2021-11-28
河南科学(2021年3期)2021-05-06
现代职业教育·高职高专(2020年1期)2020-08-16
合肥工业大学学报(自然科学版)(2020年2期)2020-03-23
南京大学学报(数学半年刊)(2020年1期)2020-03-19
计算机应用(2017年3期)2017-05-24
时代金融(2017年6期)2017-03-25
电子制作(2017年23期)2017-02-02
商场现代化(2016年11期)2016-05-20
