
遗传性压力易感性周围神经病一家系报告
2019-04-25 08:15姜方超李晓鹏张向红
中风与神经疾病杂志 2019年12期
姜方超, 马 静, 李 丹, 李晓鹏, 张向红
遗传性压力易感性周围神经病(hereditary neuropathy with liability to pressure palsies,HNPP)是由17p11.2上的PMP22基因突变引起的常染色体显性遗传性周围神经病。其以反复发作性无痛性单神经病为典型特征,多首发于10~30岁。神经电生理检查常表现为广泛性多发性周围神经潜伏期延长,传导速度减慢,易卡压部位尤其明显。周围神经腊肠样改变是其特征性病理改变,基因检测可明确诊断。在临床上因广大神经科医师对神经系统遗传病的认知缺失,常造成遗传性压力易感性周围神经病的漏诊和误诊。本文报道经基因确诊的遗传性压力易感性周围神经病一家系,对其家系成员的临床表现进行简要描述,希望能提高大家对此病的认识。
1 临床资料
先证者(Ⅳ-2),女性,16岁,拉丁舞者。因“右下肢麻木无力9 d”入院。患者9 d前上舞蹈课拉腿时突发右下肢麻木无力,主要表现为抬脚费力伴小腿前外侧及足背部麻木,行走时明显跨阈步态,患者自认为拉伤、未予诊治,近几日患者症状缓解不明显遂就诊于我院门诊,行肌电图提示“多发性感觉运动神经对称性脱髓鞘”,门诊以“多发性周围神经病”为诊断收入我科。发病前1 m内无感染史,无特殊药物服用及接触史。先证者在半年前练舞蹈时曾出现过右下肢轻度麻木无力,持续约2 w后完全缓解;其后不久在练舞时出现了右上肢轻微麻木无力,持续约2 d自行缓解。入院查体:神志清,精神可,瘦高长脸,四肢细长,脑神经正常,右下肢腓浅神经分布区痛觉减退,右下肢深感觉减退,右足及足趾不能背曲,足不能外展,四肢键反射对称减弱,高弓足,病理征末引出(双下肢查体见图1)。余神经系统检查均正常。肌电图示:双正中神经、双尺神经远端运动神经传导速度减慢、(Erb,s点-肘)运动神经传导速度正常,各段潜伏期明显延长、波幅正常伴波形离散;双侧腓总神经各段运动神经传导速度减慢(远端著)、潜伏期延长、右侧近端波幅下降;双胫神经运动神经传导速度明显减慢、近端潜伏期延长、波幅下降、远端潜伏期正常、波幅正常;右正中神经、双尺神经、双桡神经、右侧腓肠神经感觉神经传导速度明显减慢、波幅明显下降,左正中神经、左侧腓肠神经感觉神经传导速度明显减慢、波幅正常。其父叙述家族内多位近亲有周围神经病史(详见后述),先证者临床诊断为压力易感性周围神经病,给予维生素B12 500 μg/d肌肉注射和维生素B1片 10 mg 每日3次口服,进一步行遗传性压迫易感性神经病PMP22基因检测(测序+MLPA,金域),结果提示PMP22整个基因杂合缺损突变变异,明确诊断为遗传性压力易感性周围神经病。发病1 m后随访,患者右下肢麻木无力明显好转,行走时稍有异常。
家族史:该家系可追溯4代26人,其中5人发病,男性4例,女性1例,另有2人疑似患病,均为男性,呈常染色体显性遗传(家系图见图2)。先证者哥哥(Ⅳ-1),27岁。22岁当兵时时曾在剧烈运动后出现双下肢无力、行走不能,2 m后逐渐好转;26岁时右侧膝盖下部磕碰后出现右下肢麻木无力,持续1 m后好转;近1 y左手小指间断麻木伴持续性内收不能。查体:左手小指内收不能,四肢腱反射对称减弱,弓形足(见图1B),其余神经系统无阳性体征。先证者父亲(Ⅲ-1),50岁。自小蹲下稍长时间后出现双侧脚面麻木,半月后可缓解;7 y前发现右手五指内收不能。查体:右手五指内收不能,四肢腱反射对称减弱,弓形足(见图1C),其余神经系统无阳性体征。先证者爷爷(Ⅱ-1),76岁。20多岁当兵时曾出现双下肢无力、行走不能,被诊断为末梢神经炎,1 m后症状逐渐好转。先证者太爷(Ⅰ-1),已去世。自先证者父亲记事起其即表现为行走时跛行。先证者四叔(Ⅲ-9)及先证者堂弟(Ⅳ-7)曾有下肢疼痛病史(具体不详)。
2 讨 论
遗传性压力易感性周围神经病(HNPP)是一种常染色体显性遗传性周围神经病,目前国内遗传性压力易感性周围神经病的患病率尚不明确,国外研究显示遗传性压力易感性周围神经病的患病率介于0.84~16/100000之间[1]。韩国一项针对11885名新生儿进行的PMP22基因筛查显示PMP22缺失突变的携带率竟高达58.9/100000,远高于之前报道的成人患病率[1]。笔者认为两者之间的差距与遗传性压力易感性周围神经病的低诊断率有关,正如我们的这一家系中多位成员患病,部分患者甚至多次发病,但均未考虑到HNPP的可能,导致了遗传性压力易感性周围神经病患病率的严重低估。
遗传性压力易感性周围神经病常累及周围神经易卡压部位,如腓总神经的腓骨小腿处、尺神经的肘管处和正中神经的腕部等,并导致反复发作性无痛性单神经病,诱发因素常为周围神经的短时间压迫或轻微外伤[2]。但HNPP的临床表现具有显著异质性,如在本家系中可表现为反复发作性无痛性单神经病、吉兰巴雷综合征样表现、慢性尺神经病等。目前国内有关遗传性压力易感性周围神经病的报道均表现为臂丛神经损害、反复发作的周围神经病和吉兰-巴雷综合征样表现。遗传性压力易感性周围神经病首次发病多在10~30岁,男女发病机率相同[2]。有文献报道遗传性压力易感性周围神经病可晚至80岁患病[2],但笔者认为可能是诊断延误所致,而非真实发病年龄。该病多于数天至数周内完全恢复,极少遗留较严重的后遗症[2]。少数患者可有不典型表现,如非进展性单神经病、复发性位置性感觉症状、慢性感觉性多神经病、腓骨肌萎缩症样表现、喉和膈神经受累、孤立性肌肉痉挛、脚尖行走等[3~8]。
颅神经受累在遗传性压力易感性周围神经病中很少受累[9]。遗传性压力易感性周围神经病的临床表现与损伤形式显著相关,如尺神经损伤常见于睡眠压迫且症状轻微,而腓总神经损伤常见于局部外伤。除此之外,遗传性压力易感性周围神经病的临床表现还与年龄及运动形式相关,如孤立性肌肉痉挛,脚尖行走仅见于儿童[8],而臂丛神经损害和吉兰-巴雷综合征样表现常见于舞蹈、瑜伽及军人高强度训练后[5,6]。多篇报道遗传性压力易感性周围神经病患者4%~47%可出现弓形足[2,3],在该家系中多名患者出现弓形足,为诊断提供了线索。弓形足常见于腓骨肌萎缩症,目前国内各中心报道的腓骨肌萎缩症各亚型的比率差别较大,但均以CMT1A、CMTX1、CMT2A最为常见[10,11]。
有研究表明弓形足在CMT1A、CMTX1、CMT2A各亚型间的占比分别为65.2%、62.5%和11.1%[11]。因CMTX1仅见于男孩,CMT2A仅见于10岁以下的儿童[11],因此该家系先证者需考虑CMT1A和遗传性压力易感性周围神经病,因两者的临床表现及查体有明显区别,本家系临床诊断为遗传性压力易感性周围神经病。
遗传性压力易感性周围神经病的神经电生理检查具有特征性,对诊断具有提示意义。正如本文中先证者在入院时仅表现为右下肢腓浅神经病变的特征,但其肌电图却诊断提示多发性感觉运动神经对称性脱髓鞘改变,即遗传性压力易感性周围神经病患者的周围神经无论是否有临床症状都可出现神经电生理检查的异常,且病变部位多位于周围神经表浅易卡压处[12]。国内报道遗传性压力易感性周围神经病的感觉神经最易累及正中神经,其次为尺神经和腓肠神经;而运动神经最易累及腓总神经,其次为尺神经和正中神经[12],这与国外报道并不完全相同[2]。近期的一项针对30岁以下遗传性压力易感性周围神经病患者的电生理研究表明年龄仅对腓总神经的远端运动潜伏期有轻微影响[13],并提出了具有高度敏感性的诊断标准(>90%):至少一侧尺神经肘管处运动神经传导速度降低;至少一侧腓肠神经远端运动潜伏期延长;双侧正中神经远端运动潜伏期延长[13]。
遗传性压力易感性周围神经病的主要病理改变是局灶性增厚的髓鞘间隔正常髓鞘区域,沿神经纤维纵轴形似“腊肠”,故又称“腊肠样神经病”[2,3]。但因神经活检这项技术在国内尚未普遍开展,随着基因检测价格的逐渐降低及应用范围的不断扩大,目前临床上遗传性压力易感性周围神经病的确诊主要依靠基因检测。遗传性压力易感性周围神经病的致病基因为位于17p11.2的PMP22基因,PMP22基因编码一种位于周围神经表面的髓鞘蛋白,因其分子重量为22 kDa而得名[14]。PMP 22蛋白占周围神经髓鞘蛋白的2%~5%,其由4个跨膜结构域、两个胞外环和胞质内的N末端和C末端组成。PMP22蛋白的同源二聚体及PMP22蛋白与PMP0蛋白的异源二聚体在两个雪旺细胞相邻膜上的相互作用对周围神经的致密性和稳定性至关重要[15]。此外,PMP22蛋白对雪旺细胞的增殖和凋亡也有重要的调节作用[15]。周围神经髓鞘蛋白22(PMP22)基因突变是遗传性周围神经病最常见的原因,不同类型的PMP22突变可导致迥然不同的临床表型[15]。染色体17p11.2-12上包含PMP22基因的1.5 mb DNA片段的重复突变可导致CMT1A,这是CMT最常见的临床亚型,约占到CMT的50%~70%。而同一片段的大片段缺失导致遗传性压力易感性周围神经病[15]。除此之外,PMP22的各种点突变也在多种遗传性周围神经病中被发现,如遗传性压力易感性周围神经病、CMT1E、婴儿起病性髓鞘严重发育不良型周围神经病、先天性低髓鞘性周围神经病等[14,15]。
综上所述,当临床遇到年轻患者表现为反复发作性周围神经病时,若肌电图提示广泛性多发性周围神经脱髓鞘改变,应想到遗传性压力易感性周围神经病的可能,需认真询问家族史,行PMP22基因检测可明确诊断。
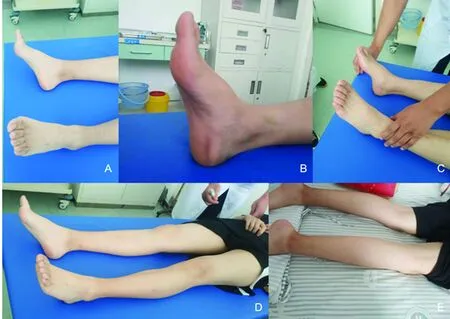
A~C:先证者(A)、先证者哥哥(B)、先证者父亲(C)均有高弓足;D:先证者双下肢细长;E:先证者入院时右下肢感觉障碍范围(笔迹)
图1 患者及其家系成员下肢表现

图2 遗传性压力易感性周围神经病一家系谱图
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
磁共振成像(2022年8期)2022-10-08
临床输血与检验(2022年3期)2022-06-22
国际检验医学杂志(2022年2期)2022-02-11
糖尿病新世界(2020年16期)2020-11-02
诊断学(理论与实践)(2020年1期)2020-04-28
党的生活(黑龙江)(2018年9期)2018-10-17
ViVi美眉(2018年5期)2018-09-18
健康大视野(2018年22期)2018-02-18
特别健康·下半月(2017年9期)2017-09-21
