
神经元核内包涵体病1例报告并文献复习
2019-04-25 08:15付胜奇张洪涛张淑玲
中风与神经疾病杂志 2019年12期
付胜奇, 宋 良, 田 芸, 沈 璐, 刘 冰, 张洪涛, 张淑玲
1 临床资料
患者,女,61岁,农民,小学文化。此次因“反复间断头痛6 y,记忆力下降1 y,行走不稳6 m,再发5 d”于2019年7月5日收入我院。患者于2013年5月无明显诱因出现头痛,以双侧眉弓、前额头痛为主,表现为胀痛,症状呈持续性,伴烦躁、易激惹,无发热、意识障碍,遂至当地医院就诊,行“腰椎穿刺术”等相关检查,考虑“脑膜炎”?给予对症治疗后(具体治疗不详)头痛症状逐渐缓解。2013年7月患者再次出现头痛、吞咽困难,头痛性质同前,就诊于“安阳肿瘤医院”,考虑“食道癌”行手术治疗,头痛给予对症治疗后症状好转出院。期间每隔2~3 m发作一次头痛,性质同前,每次自行服用止痛药物(具体药物不详)后症状可逐渐缓解。自2018年以来患者逐渐出现记忆力减退,以近记忆力下降为主,表现为不能记起刚刚发生的事情,不能保留新获得的信息,丢三落四,计算不能,但能完成日常生活劳动,如使用洗衣机、做饭等,伴便秘、尿频、尿急,未予处理。2019年1月患者再次出现头痛,全身乏力,行走不稳,就诊于当地医院,行头部CT提示“双侧白质病变伴脑萎缩”,未给予特殊处理。5 d前(2019年7月5日)患者再次出现头痛,程度较前加重,伴四肢乏力,遂至我院就诊,以“头痛原因待查”收住入院。既往患“高血压病”8 y,最高血压160/100 mmHg,服用“吲达帕胺片1片每日一次”,血压控制基本正常。患“2型糖尿病”6 y,未正规治疗及监测血糖。家族史:母亲、大哥、二哥、大姐均因“食道癌”已故。家族中无头痛等类似病史。查体:Bp:138/89 mmHg,疼痛3分。意识清楚,精神差,言语流利,远、近记忆力减退,计算力明显减退,定向力、理解力稍减退。双侧额纹对称,双侧瞳孔等大等圆,直径约2.0 mm,光反射存在,余颅神经(-)。四肢肌力5级,肌张力正常,腱反射(-),双侧巴氏征阴性。深浅感觉、共济运动未见异常。颈软,克氏征、布氏征阴性。心肺听诊未见明显异常。MMSE:21分;MOCA:14分;CDR:1.0分;ADI:23分。辅助检查:血尿粪常规、甲状腺功能6项、甲状腺抗体、血凝4项、血脂、Hcy、肝功能、肾功能、电解质、心肌酶谱、血沉、叶酸、维生素B12、BNP、h-CRP、降钙素原、血氨、血乳酸均未见明显异常。葡萄糖:7.81 mmol/L。糖化血红蛋白:8.69%。抗心磷脂抗体:阴性。传染病8项:乙肝病毒核心抗体阳性(+),乙肝病毒e抗体阳性(+),乙肝病毒表面抗体阳性(+)。肿瘤标记物:甲胎蛋白、CEA、CA199、CA125、CA153、CA724、CY211、SCC、NSE均未见异常。副肿瘤抗体均阴性。风湿免疫指标:类风湿因子、抗链“O”、抗核抗体、抗蛋白酶3抗体、抗ENA抗体、免疫球蛋白3项、抗角蛋白抗体、RA33、抗环瓜氨酸肽均阴性。脑脊液压力120 mmH2O,CSF常规:潘氏实验(+),生化:PRO:115.05 mg/dl,结核细菌涂片、细菌培养、抗酸染色、ADA、墨汁染色、AQP4、自身免疫性脑炎6项、寡克隆区带均阴性。头部MRI+MRA+CEMRA示(见图1):双侧额叶、顶叶、颞叶可见白质异常信号影,DWI 成像可见双侧额、顶、颞叶沿皮髓质交界处呈绸带状高信号。泌尿系超声:膀胱残余尿量86 ml。四肢肌电图:左腓总神经MCV 减慢,波幅下降;双尺神经、双正中神经、双胫神经、右腓总神经MCV 减慢,波幅正常;SCV:右桡神经、双腓浅神经SNAP 未引出;双尺神经、右正中神经、左桡神经SCV 减慢,波幅正常;提示四肢多发性周围神经损害。肛门括约肌肌电图未见异常。脑电图未见异常。留取左侧小腿处距外踝约10 cm的皮肤进行活检,病理检查提示(见图2):小汗腺细胞、成纤维细胞、脂肪细胞内可见核内包涵体。基因检测提示NOTCH2NLC 基因5’端的GGC异常重复扩增[1]。NIID的诊断流程图(见图3)。入院后给予止痛、改善认知功能、控制血压、血糖等对症支持治疗,好转出院。
2 讨 论
该患者中年女性,慢性进展性起病,主要临床表现为3点:(1)中枢神经系统受累:包括头痛、认知功能减退、精神行为异常、行走不稳;(2)周围神经症状:肌电图提示多发周围神经损害;(3)自主神经症状:患者出现便秘、尿频、尿急,查体可见双侧瞳孔缩小,膀胱残余尿量为86 ml。影像学表现MRI T2、Flair可见广泛的脑白质病变,DWI上可见额叶、颞叶沿皮质、髓质交界处呈飘带状高信号。皮肤活检发现小汗腺细胞、成纤维细胞、脂肪细胞内可见核内包涵体。根据患者既往病史,临床症状、体征及辅助检查,诊断此患者为散发型成人型神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)。神经元核内包涵体病,也被称为神经元在细胞核内的嗜酸性透明质疾病(neuronal intranuclear hyaline inclusion disease,NIHID),它是一种进展缓慢的神经退行性疾病,其特征性表现是在中枢和外周神经系统以及内脏器官中存在有嗜酸性透明质核内包涵体。
1968年由Lindenberg在1例临床表现为进行性痉挛、精神发育迟滞、共济失调和自主神经功能损害的28岁患者的大脑和内脏细胞内发现了核内包涵体,首次提出“神经元核内包涵体病(NIID)”的诊断[2]。此后NIID的发展史分为两个阶段,第一阶段从1968年到2011年,通过尸检、神经活检、直肠活检等途径报道了大约40余例的NIID患者,绝大多数患者均为婴幼儿或者青少年起病,主要表现为大脑皮质功能障碍和椎体外系症状为主的神经系统变性病变[3,4]。第二阶段从2011年至今,2011年由日本学者Sone等采用皮肤活检成功诊断了NIID疾病[5]。2014年Sone在NIID患者的磁共振影像中发现了特征性的改变即DWI成像显示皮质-髓质交界高信号病变,可以作为诊断此病的另一个重要线索[6,7]。因此,目前认为NIID的诊断主要通过磁共振成像联合皮肤活检来确诊。
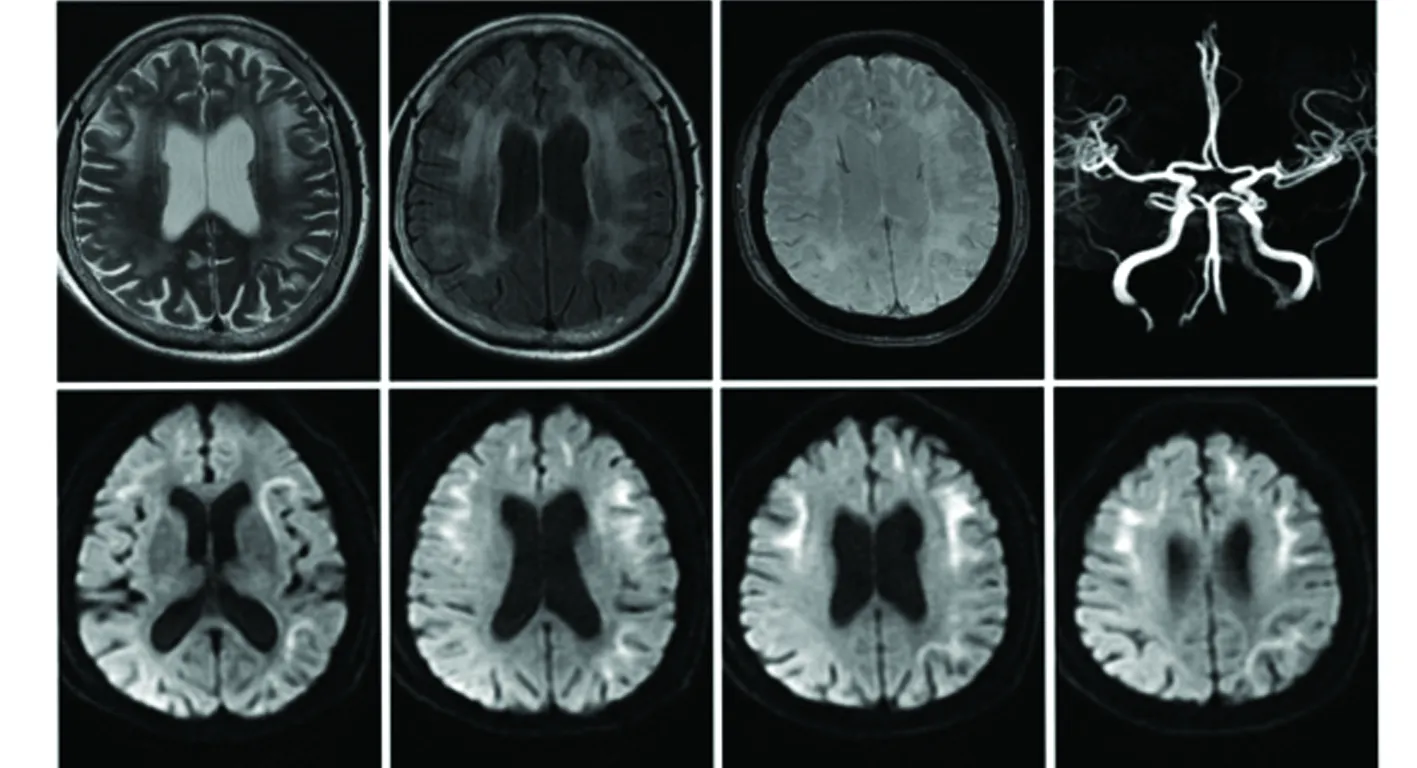
图1 T2、Flair成像可见双侧额叶、顶叶、颞叶可见白质异常信号影, DWI 成像可见双侧额、顶、颞、枕叶沿皮髓质交界处绸带状高信号影

图2 基因检测提示:NOTCH2NLC 基因5’端的 GGC 异常重复扩增。病理检查提示:小汗腺细胞、成纤维细胞、脂肪细胞内可见核内包涵体

图3 NIID诊断流程图
目前,关于NIID的病理生理机制尚未明确,NIID是遗传性神经变性疾病中的一种重要病理表现。其组织细胞中存在嗜酸性透明包涵体,而此包涵体是位于核周直径为1.5~10 μm的圆形物质,泛素、p62阳性,且由电镜下无模结构的纤维物质组成[6]。尸检研究发现[8],嗜酸性透明包涵体主要存在于神经元和神经胶质细胞中,并且神经胶质细胞占大多数比例,这种嗜酸性透明包涵体主要分布在3个系统包括中枢、周围神经系统和非神经组织中。大部分神经变性疾病是由基因突变表达的三核苷酸重复疾病蛋白引起,并且所有已知的三核苷酸重复疾病神经元内都存在神经元核内包涵体,故目前对神经元核内包涵体的研究主要表现在此类疾病中[9]。Riess[10]在动物研究发现,神经元核内包涵体形成的必要条件是特异性基因扩增的三核苷酸重复序列,其主要是通过泛素/蛋白水解酶途径在包涵体的形成中发挥重要作用。有研究指出嗜酸性核内包涵体主要存在于患者的脂肪细胞、成纤维细胞和皮肤组织中的汗腺细胞中[5]。因此,皮肤活检中特征性核内包涵体的存在可以作为NIID的主要诊断标准。
NIID的临床症状主要分类及3大系统,包括中枢、周围及自主神经系统。其临床特征多种多样,从婴儿到老年均可发病,根据发病年龄分为未成年型和成年型,未成年型分为儿童型和青少年型;成年型分为散发型和家族型[3]。(1)散发型NIID:平均发病年龄为63.6岁,病程平均持续时间5.3 y[11],其主要临床表现常以痴呆为主,还可以表现为精神行为异常、共济失调、全面强直阵挛发作、发作性意识障碍、亚急性发作性脑炎、强直、震颤、肌无力、感觉障碍及自主神经障碍。(2)家族型NIID的临床症状往往长达数十年,主要分为痴呆组和肢体无力组两组。痴呆组的平均发病年龄为56.2岁,平均持续时间7.6 y,以痴呆(100%)为主,伴有轻微的周围、中枢及自主神经症状;而肢体无力组的平均发病年龄为27.5岁,病程平均持续时间为21.1 y,以肢体无力(100%)为首发症状,其余可出现感觉障碍、呕吐、膀胱功能障碍等。本研究患者系散发型,临床表现主要累及中枢、周围及自主神经系统,为散发型NIID常见的临床症状。
磁共振是诊断评估疑似神经退行性疾病的重要工具,特别是在早期临床症状和体征呈现非特异性[12]。NIID患者的头部MRI T2及Fliar成像可见广泛脑白质病变,在T2成像上均可见Fazekas分级评分2级以上的脑白质病变,部分患者病变可累及到胼胝体、外囊、小脑中叶、丘脑及基底神经节等。所有的患者均可出现广泛性脑及小脑萎缩等改变[13]。另外,DWI成像上均可见皮质下灰质与白质交界处明显的曲线性高信号,此高信号仅存在于皮质、髓质交界处,主要累及额叶、顶叶和颞叶,并随着疾病的进展病灶不断向后延伸至枕叶。这种特征性的DWI高信号被命名为皮质下绸带征,为诊断NIID提供了强有力的线索。因此,当患者出现特征性的临床表现,结合皮质髓质交界区DWI高信号时,提示临床医生应该疑诊NIID,建议进一步行皮肤活检病理检查,明确NIID的诊断。
NIID是一种神经系统变性病,随着诊断的明确,目前尚无明确有效的药物来治疗,期待着越来越多的病例被发现,治疗方法能够有所进展。目前对于该病的治疗,主要以对症支持治疗为主,如头痛给予对症止痛药物、癫痫发作给予抗癫痫治疗、周围神经受累给予营养神经治疗等。
综上所述,成人发病的NIID似乎不是一种非常罕见的疾病,其患病率可能比以前认为的要高,并且NIID的诊断可能被低估。因此,当T2加权像上出现广泛的脑白质病变,DWI成像上出现皮质下绸带征时,需从影像中快速识别NIID患者。神经内科及放射科医师都应该认识到此典型的磁共振影像表现,结合临床症状及皮肤活检,来进一步确诊NIID。通过对临床上不断增多的新的病例的总结,期待更多的有关NIID的发病机制及治疗方面的研究。
猜你喜欢
昆明医科大学学报(2021年3期)2021-07-22
电子产品世界(2021年8期)2021-01-16
医学新知(2019年4期)2020-01-02
保健与生活(2019年15期)2019-09-12
中国计算机报(2019年49期)2019-02-07
中国疼痛医学杂志(2019年9期)2019-01-04
健康女性(2016年11期)2017-02-14
创新时代(2016年8期)2016-10-21
经营者·汽车消费报告(2013年11期)2014-01-23
美文(2009年10期)2009-06-04
