
比较两种转录激活系统对生殖相关基因的表达调控
2019-04-20 08:17卢佳豪黄亚萍肖亚梅赵小阳
生命科学研究 2019年5期
卢佳豪,黄亚萍,罗 芳,肖亚梅,赵小阳*
(1.湖南师范大学生命科学学院省部共建淡水鱼类发育生物学国家重点实验室,中国湖南长沙410081;2.南方医科大学基础医学院发育生物学教研室,中国广东广州510515)
规律成簇间隔短回文重复(clustered regularly interspaced short palindromic repeats,CRISPR)是细菌和古生菌细胞保护自身不受病毒侵害的防御系统[1]。当微生物遭到病毒入侵时,CRISPR RNA通过互补配对结合到病毒基因组,并通过核酸酶Cas9特异性地切割病毒DNA,以实现自我保护[2]。近年来,大量研究应用CRISPR-Cas9技术在人、小鼠、猴、斑马鱼和拟南芥等多个物种上进行了基因敲除,此外研究者们通过突变Cas9的两个核酸酶结构域HNH和RuvC,产生没有切割活性但在sgRNA(single guide RNA)的引导下仍能特异性识别并结合靶向序列的dCas9(dead-Cas9)[3]。dCas9可与不同功能的蛋白质结构域融合,从而实现定向的基因转录调控(包括转录激活及转录抑制)、表观遗传学修饰以及染色体可视化等方面的应用。其中,CRISPR介导的转录激活系统(CRISPR activation,CRISPRa),是将dCas9与转录激活因子或组蛋白修饰酶的核心结构域进行融合来实现基因的靶向激活[4]。
最初,CRISPRa系统是将dCas9与多个串联的VP16转录激活域(如VP64、VP160等)融合来激活内源性基因表达,但效率较低[5]。随后,研究者们尝试筛选不同转录激活复合物来优化系统,统称为二代激活系统,包括由VP64、p65AD和巴尔病毒Rta反式激活因子组成的VPR系统及MS2、p65和HSF1转录激活蛋白质组成的SAM系统[3]。此外,还有团队将Cas9与人乙酰转移酶p300的催化核心结构域进行融合,构成Cas9-p300系统,该系统通过催化靶基因启动子或增强子上的H3Lys27实现乙酰化,使得靶基因染色质结构变得疏松,进而招募转录激活因子来激活靶基因的表达[3]。二代激活系统提高了转录激活效率,因此近年来不断有研究者应用CRISPRa来实现细胞定向分化或重编程。此外,有研究者通过使用CRISPRa系统激活内源性Fst(follistatin)基因的表达,在一定程度上缓解了肌营养不良症的小鼠肌肉萎缩的表型[6],这表明CRISPRa系统的基因激活在疾病模型及治疗方面具有应用潜力。
本文旨在比较Cas9-p300与dCas9-VPR两个系统对生殖发育相关基因的激活效率,这将为研究生殖细胞分化及未来利用转录激活系统实现细胞命运转变提供基础,也将为其他发育系统的基因功能研究或定向分化应用提供借鉴。
1 材料与方法
1.1 细胞
人肾上皮细胞293T为本实验室所有。感受态细胞DH5α和stbl3购自深圳康体生命科技有限公司。
1.2 载体
载体p2U6-Cas9-p300-mCherry为本实验室所有。载体PB-TRE-dCas9-VPR(#63800)、MLM-3636(#43860)购自 Addgene 官网(http://www.addgene.org/)。
两个转录激活系统的载体分别是p2U6-Cas9-p300-mCherry和 PB-TRE-dCas9-VPR(图 1),其中Cas9-p300系统由野生型Cas9与乙酰转移酶p300形成融合蛋白。

图1 载体结构示意图以及载体构建示意图(A)p2U6-Cas9-p300-mCherry载体结构示意图;(B)PB-TRE-dCas9-VPR载体结构示意图;(C)MLM3636载体结构示意图。Fig.1 Schematic of transcription activation systems(A)p2U6-Cas9-p300-mCherry system;(B)PB-TRE-dCas9-VPR system;(C)MLM3636 system.
1.3 试剂
限制性内切酶BsmB I(#R0613L,美国NEB公司);T4连接酶(#M0202V,美国NEB公司);质粒小提试剂盒(DC201,南京诺唯赞生物科技有限公司);DNA纯化回收试剂盒(DC301,南京诺唯赞生物科技有限公司);细胞转染试剂Peimine(ab-143678,英国Abcam公司);Trizol(R401-01,南京诺唯赞生物科技有限公司);HiScript Q RT Super-Mix for qPCR(gDNA wiper)(R123-01,南京诺唯赞生物科技有限公司);A301 2×RealStar Green Fast Mixture(A301-10,北京康润诚业生物科技有限公司)。
1.4 方法
1.4.1 细胞培养
将293T细胞接种于含10%胎牛血清的DMEM培养液中,在37℃、5%CO2的恒温培养箱进行培养。
1.4.2 sgRNA设计
根据CRISPR/Cas9原理中sgRNA设计原则,将目的基因1号外显子上游promoter 200 bp区域序列提交至在线设计sgRNA的网站(http://chopchop.cbu.uib.no/),待系统生成推荐序列后选择高评分序列作为备选sgRNA,详细信息见表1。
1.4.3 重组表达sgRNA载体
BsmB I酶切后回收的MLM3636载体骨架与sgRNA退火后的双链DNA(dsDNA)利用T4连接酶进行连接反应,连接产物转化到DH5α感受态细胞中。将转化好的感受态细胞涂布于含氨苄青霉素(ampicillin)的LB培养基标准平板上。挑取单克隆进行菌液PCR鉴定,将PCR检测阳性的克隆加入含氨苄青霉素的LB液体培养基中进行连续培养,12 h后提取质粒,送往北京睿博兴科生物技术有限公司进行核酸检测,查看sgRNA是否正确整合进入载体。
1.4.4 转染293T细胞
当293T细胞密度达到50%时,利用 PEI(polyethylenimine)法共转染转录激活系统载体与sgRNA的载体,从而激活目的基因。具体而言,在分别转染两种不同的转录激活载体p2U6-Cas9-p300-mCherry和PB-TRE-dCas9-VPR的同时,转染带有相同sgRNA的载体MLM3636-sgRNA,由此比较这两种转录激活系统对同一个基因的激活效率。另外,设置对照组为仅转染转录激活系统载体的293T细胞。
1.4.5 总RNA提取与逆转录
收取转染72 h后的293T细胞,用Trizol法提取RNA。利用诺维赞HiScript Q RT SuperMix for qPCR(gDNA wiper)试剂盒对提取的RNA进行逆转录。每个RNA样本取1 μg总量进行逆转录,生成cDNA。
1.4.6 实时荧光定量PCR
以合成的cDNA为模板,使用A301 2×RealStar Green Fast Mixture对样本进行表达检测。每次定量分析设3个重复样品,PCR引物信息见表2。以GAPDH为内参基因,以no gRNA组为对照样本,进行归一化处理,对比Cas9-p300和dCas9-VPR系统的激活效率。实验结果以平均值±标准误差表示。组间比较利用GraphPad Prism 6软件进行t-test分析。无显著差异(NS)为P>0.05,显著性差异为P<0.05,极显著性差异为P<0.01。

表1 基因靶点序列Table 1 The target sequences of genes

表2 实时荧光定量PCR引物序列Table 2 The primer sequences for real-time quantitative PCR assay
2 结果
2.1 靶向目的基因启动子区域的sgRNA载体构建
由于本文应用的转录激活系统Cas9-p300是利用野生型Cas9与乙酰转移酶p300形成的系统,在长度为20个碱基的sgRNA引导下会对DNA双链进行切割,而14~15个碱基的 sgRNA(短gRNA)能够引导Cas9到靶位点且阻止DNA双链断裂[6]。因此,我们构建长度为14 bp的sgRNA对目的基因进行激活(图2A)。
首先合成两条互补的oligo序列,并在正义链5'端添加ACAC,反义链 5'端添加AAAA作为黏性末端,随后将其退火形成互补的DNA双链。将退火的双链DNA连接到通过限制性内切酶BsmBⅠ线性化的载体MLM3636,从而形成携带sgRNA的重组载体(图2A)。重组载体经菌落PCR鉴定及进一步的测序分析证实,sgRNA均正确插入到MLM3636载体,说明靶向目的基因启动子区域的sgRNA载体构建成功(图2B~E)。

图2 sgRNA载体构建示意图以及部分载体测序结果(A)载体MLM3636-gRNA构建流程图;(B)~(E)针对目的基因NANOS2设计的sgRNAs(NANOS2-1~NANOS2-4)及其所构建载体的测序结果。Fig.2 Schematic of sgRNA vector construction and partial vector sequencing results(A)The construction process of recombinant plasmid MLM3636-gRNA;(B)~(E)sgRNAs(NANOS2-1~NANOS2-4)designed for target gene NANOS2,and the sequencing results of the constructed vectors containing above sgRNAs.
2.2 转录激活系统高效转染293T细胞
我们针对 BLIMP1、TFAP2C、DDX4、NANOS2、SOX17和DAZL 6个基因分别设计了多个sgRNA,并将sgRNA载体与p2U6-Cas9-p300-mCherry或PB-TRE-dCas9-VPR载体共同转染入293T细胞内。由于p2U6-Cas9-p300-mCherry载体携带红色荧光标记蛋白质mCherry,所以我们能通过荧光体视显微镜观测到红色荧光的发光效率并能以此监测PEI法转染293T细胞的实时效率。结果显示,利用PEI转染法转染p2U6-Cas9-p300-mCherry转录激活系统的效率高达90%以上(图3)。
2.3 实时荧光定量PCR比较两个系统对不同基因的激活效率
为比较两种转录激活系统的激活效率,我们收取了转染72 h后的293T细胞并且提取了RNA。将RNA逆转录成cDNA后,通过实时荧光定量PCR检测目的基因的表达水平,结果如图4所示。
荧光定量PCR结果显示,Cas9-p300与dCas9-VPR能够上调5个基因的表达,分别是BLIMP1、TFAP2C、NANOS2、SOX17 和 DAZL,但对 DDX4基本无效。需要指出的是,Cas9-p300系统对BLIMP1基因表现出更高的激活效率。其中sgRNA-1和sgRNA-2对应的激活效率约为108倍,而dCas9-VPR系统的激活效率约为50倍,差异均具有显著性(P<0.05);但是在BLIMP1 sgRNA-3(P<0.05)与 sgRNA-5(P<0.01)诱导下,dCas9-VPR系统的激活效率比Cas9-p300系统更高。其次,dCas9-VPR 系统在激活 NANOS2、SOX17、DAZL这3个基因时比Cas9-p300系统更为有效(P<0.05)。两个系统在sgRNA-4诱导下对TFAP2C均达到最高激活效率,并且两者之间无显著差异。而g1~g3所对应的相对表达水平虽具有统计学差异,但两者对目的基因TFAP2C的激活倍数太低,无法被认定为有效激活。关于两个系统对TFAP2C激活效率都不高的情况,我们推测可能与该基因本底表达较高[5]有关。综上所述,Cas9-p300系统与dCas9-VPR系统对不同基因或同一基因选用不同的sgRNA,所产生的激活效率会有所不同。
3 讨论

图3 sgRNA与Cas9-p300系统共转染293T细胞48 h后的荧光图BF 表示 293T 细胞的白光图(标尺:200 μm)。Fig.3 Fluorescence images of 293T cells after transfection 48 h with sgRNA and Cas9-p300BF represents bright field image of 293T cells(scale bar:200 μm).
之前的研究表明,利用第二代转录激活系统VPR、SAM和Suntag在293T细胞中对相同的6个目的基因进行激活时,SAM系统在提供高效率的基因诱导方面最为稳定,而且与其他两种系统的差异不会大于5倍;VPR和Suntag系统则在特定的一些基因中表现出比其他系统更为高效的激活效率[7~8]。由于每个激活系统具有各自不同的原理和特点,因而在对一些特异性基因进行激活时尚不能明确哪种系统是最有效的[9~10]。
本研究利用Cas9-p300与dCas9-VPR两种不同的转录激活系统,对生殖细胞特化相关基因的启动子区域进行内源性激活,比较了在相同gRNA的引导下Cas9-p300与dCas9-VPR转录激活系统在293T细胞中的激活效率。结果表明,在对生殖细胞特化相关基因进行激活时,激活效率呈现出以下3种模式:1)Cas9-p300系统更高效(BLIMP1);2)dCas9-VPR系统的效率更高(NANOS2、SOX17、DAZL);3)两个系统的激活效率相近。我们推测造成激活效率不同的原因可能是不同基因的染色质开放状态不同,这使得作用方式不同的系统产生不同的激活效率。上述结果说明,不同的转录激活系统在激活生殖细胞特化基因时,具有不同的效率,这为实现靶基因的激活提供了候选策略。由此,本研究为后续研究生殖细胞分化铺垫了基础,也为利用转录激活系统研究细胞命运转变提供了借鉴。
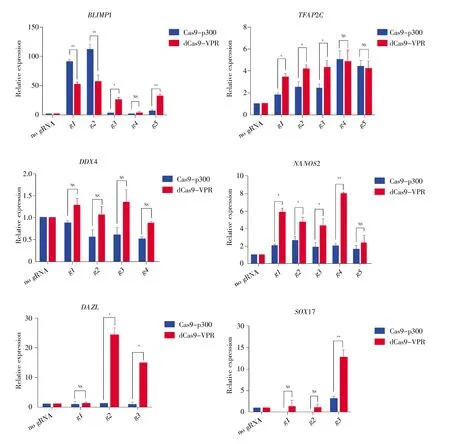
图4 两个系统对不同基因的激活效率分析横坐标表示不同的gRNA,纵坐标表示该gRNA对目的基因表达水平的影响。**:P<0.01;*:P<0.05。Fig.4 Activation efficiencies of the two systems for expression of different genes in 293T cellsThe horizontal coordinates denote different gRNAs,and the longitudinal coordinates represent the corresponding expression levels of target genes.**:P<0.01;*:P<0.05.
猜你喜欢
华人时刊(2022年9期)2022-09-06
昆明医科大学学报(2021年8期)2021-08-13
华人时刊(2020年15期)2020-12-14
甘肃教育(2020年14期)2020-09-11
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
时代英语·高二(2015年1期)2015-03-16
中国卫生(2014年11期)2014-11-12
中国火炬(2013年11期)2013-07-25
中国火炬(2013年10期)2013-07-24
中国洗涤用品工业(2011年6期)2011-03-20
