
抗流感药法匹拉韦杂质的合成
2019-04-12 05:32邓玉晓段崇刚林治秘任业明孙晋瑞
食品与药品 2019年2期
邓玉晓,段崇刚,林治秘,李 丹,任业明,孙晋瑞
(山东省药学科学院 山东省化学药物重点实验室,山东 济南 250101)
法匹拉韦(favipiravir,商品名为Avigan),化学名为6-氟-3-羟基吡嗪-2-甲酰胺,是由日本富士胶片旗下富山化学工业公司研发的一种新型流感治疗药物,2011年3月在日本完成Ⅲ期临床试验,2014年3月被日本政府批准上市[1]。2014年3月,日本厚生劳动省批准其用于治疗新型和复发型流感[2]; 2015年7月13日被台湾批准上市用于治疗流感;2016年6月14日被几内亚批准用于治疗埃博拉病毒[3]。本品为病毒RNA聚合酶抑制剂,能选择性阻断病毒RNA的合成,对哺乳动物细胞内的RNA合成不会产生任何抑制作用,因此,是一种安全有效的抗病毒药物[4]。由于其特定的作用机制,除流感病毒外,法匹拉韦还可对抗其他多种RNA病毒,如HIV、黄热病、SARS、埃博拉等[5]。
目前,文献[6-8]报道的路线中,具有工业化前景的为3,6-二氯吡嗪-2-甲腈(化合物2)在相转移催化剂四丁基溴化铵(Bu4N+Br-)的催化下,与无水氟化钾发生双氟取代反应,得3,6-二氟吡嗪-2-甲腈(化合物3);再与醋酸和三乙胺作用生成6-氟-3-羟基吡嗪-2-甲腈(化合物4);最后在浓硫酸作用下水解生成法匹拉韦。合成路线详见图1。

图1 法匹拉韦的合成路线
笔者查阅了大量文献,均未检索到法匹拉韦杂质相关报道,为了控制法匹拉韦的质量,必须建立严格的质量标准,经大量的实验探索和求证,最终确证了两个杂质:6-氯-3-羟基吡嗪-2-甲酰胺(杂质1)和6-氟-3-羟基吡嗪-2-甲酸(杂质2),同时研究了相关杂质的制备方法,以为该产品的相关技术人员提供参考。

图2 法匹拉韦合成中两种主要杂质
从化学结构与分子活性角度分析,化合物2在3位和6位均有一个氯原子取代,然而,与两个氯原子直接相连的碳原子的亲电活性却相差较大,3位碳原子由于受到芳香环上相邻氮原子和邻位氰基的双重吸电子作用,电子云密度较低,极易受到氟离子的进攻,发生氟代反应; 6位碳原子受到芳香环上相邻氮原子的吸电子作用,而氰基却位于其间位,对其吸电子作用较小,电子云密度相对较高,所以,在相同条件下,有可能会产生3位氟代反应完全,而6位氟代反应不完全的情况,即生成3-氟-6-氯吡嗪-2-甲腈(化合物5),该化合物在冰醋酸和三乙胺作用下生成6-氯-3-羟基吡嗪-2-甲酰胺(化合物6),最后在浓硫酸作用下生成杂质1。由于化合物3中副产物5的含量较低,且在法匹拉韦原料药合成过程中转化为杂质1,并未在法匹拉韦原料药中检测到化合物5和化合物6。杂质1的来源流程图详见图3。化合物4中2位氰基在酸性或碱性条件下水解,通过调节浓硫酸的浓度和反应温度可控制反应主产物为法匹拉韦,但是过度水解产物杂质2却很难避免,这一点在相关文献[9]中已有详尽报道。

图3 杂质1的来源流程图
1 合成路线
1.1 杂质1的合成路线
相关文献[10-11]报道了杂质1的合成方法,即以6-氯-3-羟基吡嗪-2-甲酸甲酯为起始原料,通过酯的氨解得杂质1。但酯的氨解反应速度慢且反应进行不彻底,所得产物中必然包含未反应完全的原料,且由于反应原料和产物极性相近,纯化困难。合成路线详见图4。

图4 文献[10-11]中杂质1的合成路线
本文采用3,6-二氯吡嗪-2-甲酰胺为起始原料,经亲核取代反应、水解反应得到杂质1。起始原料易得,且反应条件温和,产物易分离纯化。详见图5。

图5 本文杂质1的合成路线
1.2 杂质2的合成路线
笔者查阅大量文献,未发现杂质2的相关报道。我们以化合物4为起始原料,采用氢氧化钠水溶液水解,然后用盐酸调节反应液pH<3,即得杂质2。
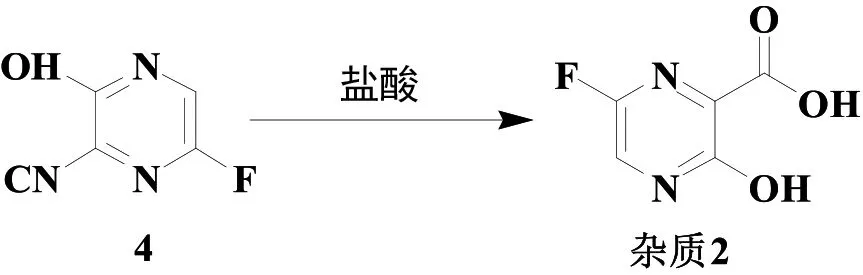
图6 杂质2的合成路线
2 仪器与材料
2.1 仪器
Varian INOVA-300 核磁共振波谱仪(美国瓦里安公司);4000 Q TRAP 质谱仪(美国AB SCIEX公司);IKA HB 10 旋转蒸发仪(德国IKA);循环水式多用真空泵(郑州长城);DZF型真空干燥箱(北京市永光明医疗仪器厂);Agilent 1100高效液相色谱仪(美国安捷伦)。
2.2 药品与试剂
3,6-二氯吡嗪-2-甲腈(四川南部县诚信公司,纯度>98.5%);6-氟-3-羟基吡嗪-2-甲腈(四川南部县诚信公司,纯度>98.5%);二氯甲烷(天津富宇,分析纯);浓硫酸(国药集团,分析纯);无水碳酸钾(国药集团,分析纯);N,N-二甲基甲酰胺(国药集团,分析纯);乙酸乙酯(天津科密欧,分析纯);二甲苯(天津富宇,分析纯);无水乙醇(莱阳经济技术开发区精细化工,分析纯);氢氧化钠(国药集团,分析纯);无水硫酸钠(国药集团,分析纯);浓盐酸(国药集团,分析纯);石油醚(天津富宇,分析纯,沸点60~90 ℃)。
3 方法与结果
3.1 6-氯-3-羟基吡嗪-2-甲腈(化合物6)的合成
取化合物2(5.0 g,0.029 mol)置入100 ml四口瓶,加无水碳酸钾(7.94 g,0.057 mol),N,N-二甲基甲酰胺(25 ml),搅拌下油浴加热,保持体系内温度50~60 ℃,反应5 h。薄层色谱(TLC)显示反应完全后,将反应液加入100 ml水中,用乙酸乙酯(100 ml×3)萃取,合并有机相,用水(100 ml×3)洗涤,经无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,得黄色油状物(4.13 g,收率:92.5%)。
3.2 6-氯-3-羟基吡嗪-2-甲酰胺(杂质1)的合成
向50 ml四口瓶中加入化合物6(4.13 g,0.027 mol),浓硫酸(10 ml),搅拌下油浴加热,保持体系内温度55~65 ℃,反应1 h。TLC显示反应完全后,将反应液加入冰水(50 ml)中,用二氯甲烷(50 ml×3)萃取,合并有机相,用水(50 ml×3)洗涤,经无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,得黄色固体(4.32 g,收率:93.6%),为杂质1粗品。向100 ml四口瓶中加入杂质1粗品(4.32 g),无水乙醇(80 ml),搅拌下油浴加热,保持回流约0.5 h,关闭加热和搅拌,自然降至室温,抽滤,得淡黄色针状结晶(3.70 g,收率:85.7%)。mp:200.5~201.6 ℃(文献[12]mp:200~202 ℃),HPLC纯度:99.6%。ESI-MSm/z:172.6[M-H]-。1H NMR(300 MHz DMSO-d6):8.61(s,2H),8.83(s,1H),13.66(s,1H);13C NMR(300 MHz DMSO-d6):169.5,157.9,142.5,140.3,128.4。
3.3 6-氟-3-羟基吡嗪-2-甲酸(杂质2)的合成
向100 ml四口瓶中加入化合物4(5 g,0.036 mol),4 mol/L盐酸(20 ml),搅拌0.5 h,搅拌下油浴加热,保持回流约3 h。TLC显示反应完全后,将反应液加入冰水(50 ml)中,过滤,滤饼采用蒸馏水(50 ml×3)洗涤,60 ℃真空干燥至恒重,得棕色粉末(4.01 g,70.4%),为杂质2粗品。向100 ml四口瓶中加入杂质2粗品(4.01 g),石油醚(40 ml),室温下打浆0.5~1 h,抽滤,得淡黄色粉末(3.87 g,收率:96.5%)。mp:178.8~180.2 ℃,HPLC纯度:99.7%。ESI-MSm/z:157.0[M-H]-。1H NMR(300 MHz DMSO-d6):8.50(s,1H),13.7(s,1H);13C NMR(300 MHz DMSO-d6):172.5,161.2,142.3,138.2,127.0。[杂质1和杂质2的纯度测定方法均为HPLC面积归一化法,色谱条件:色谱柱:Intersil ODS-3,流动相:V缓冲溶液:V甲醇=45:55,缓冲溶液配置方法:0.5 g磷酸二氢钾,1.0 g磷酸二氢钠,加水1000 ml,摇匀即得,流速:1 ml/min,柱温:30 ℃,进样体积:20 μl,运行时间:40 min,检测波长:238 nm]。
4 讨论
4.1 本研究杂质1和杂质2的合成所采用的起始原料易得,反应条件温和,未涉及高温高压超低温等苛刻反应条件,反应的可操作性高。
4.2 采用重结晶或打浆的方法对2个杂质提纯,收率高,操作简便,避免了硅胶柱纯化等繁琐操作步骤。
4.3 本研究合成的2个杂质均纯度高,性状好,易于保存。
5 结论
本文分别以3,6-二氯吡嗪-2-甲腈和6-氟-3-羟基吡嗪-2-甲腈为起始原料,合成了法匹拉韦的两个杂质,并通过MS、1H NMR、13C NMR等方法进行了结构确证。解决了2个杂质对照品来源问题,对法匹拉韦的质量控制有重要意义。
猜你喜欢
中国防痨杂志(2022年1期)2022-11-24
世界农药(2022年10期)2022-11-10
当代化工研究(2022年11期)2022-06-27
能源化工(2021年2期)2021-12-30
实用药物与临床(2021年8期)2021-01-08
农药科学与管理(2019年10期)2019-04-20
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
应用化工(2015年12期)2015-04-14
中国酿造(2015年4期)2015-01-26
