
人参皂甙Rb1通过调控缝隙连接蛋白40治疗脑缺血再灌注损伤的机制
2019-04-11 07:59任大斌童武松冯九庚
中风与神经疾病杂志 2019年3期
施 虹, 任大斌, 郑 平, 童武松, 冯九庚, 段 剑, 陈 伟
脑缺血再灌注损伤(I/R)所带来的致死及致残率仍然较高[1],目前临床上缺乏明确有效的治疗手段。人参皂甙(GS)是人参中最主要的活性物质,已经被证明对神经损伤类疾病有着明显的保护作用[2]。GS-Rb1已被证明是GS中最主要的成份。我们的前期研究显示:GS-Rb1的脑保护作用可能和下调缝隙连接蛋白(Connexin,Cx)40有关[3],但尚未进一步证明相关的分子机制。本研究通过缺血再灌注损伤后调控PKA途径,观察PKA途径参与GS-Rb1下调Cx40表达的相关性,旨在探讨GS-Rb1对脑I/R损伤保护作用的具体机制及可能涉及多信号分子信号传导途径,为脑缺再灌注损伤的分子水平的治疗奠定基础。
1 材料和方法
1.1 实验动物、试剂及仪器 100只3月龄、体重350~450 g的雄性SD大鼠购置于上海毕凯实验动物有限责任公司(沪ICP备05033115)苯巴比妥麻醉钠购置Fluka公司(德国)。GS-Rb1(纯度大于99%)购自上海邦景(中国),H-89(纯度大于99%)购自上海RBI生物(中国),兔抗大鼠Cx40一抗购自Santa Cruz(美国),兔抗大鼠GAPDH一抗购自Abcam(英国),辣根酶标记二抗均购自Abcam (英国),化学发光试剂盒购自中山金桥生物科技(中国),所有ELFA、ELISA试剂盒均购自武汉博士德生物(中国),激光多普勒血流仪购自Stockholm(瑞典),蛋白电泳仪购自Bio-Rad(美国),NAPDH氧化酶活性试剂盒购自Beyotime (中国),M200Pro免疫酶标仪购自TECAN(奥地利)。
1.2 动物模型制作 实验用大鼠在通风房间内按照光照:黑暗时间比例为12∶12 h,给予水和食物适应性喂养1 w,动物伦理已得到上海市浦东新区人民医院实验动物伦理委员会的同意。按照夹闭法建立大鼠缺血模型:将3%戊巴比妥以65 mg/kg的浓度给予大鼠麻醉后,以颈部正中切口进入,找到双侧颈总动脉,分离血管外膜及迷走神经,用微血管夹将双侧颈总动脉夹闭,整个手术过程用动物实验专用体温装置把动物体温维持的37 ℃摄氏度。夹闭效果用激光多普勒血流仪进行测量(以小于正常血流量的25%作为有效)。阻断双侧颈总动脉血流30 min后,撤去血管夹,恢复血流,产生再灌注(以血流量恢复至基线水平75%视为有效)。
1.3 药物的干预及分组 100只SD大鼠被随机分为①假手术(sham)组、②灌注后GS-Rb1(I/R+GS-Rb1)治疗组、③灌注后GS-Rb+H-89阻断(I/R+GS-Rb1+H-89)组、④溶媒(I/R+DMSO)组及⑤损伤(I/R)组,每组20只。①仅暴露而不夹闭颈总动脉,②③均采用夹闭颈总动脉法制作脑I/R模型,1 h后腹腔注射GS-Rb1及GS-Rb1+H-89,④为I/R形成1 h后腹腔注射DMSO,而⑤则是I/R形成后不给予处理,所有组实验时间均设定为I/R后8 h。每组取10动物行行为学检测和脑组织含水量测定,另10只动物则行蛋白学检测。根据我们之前预实验及前期实验显示,为获得P值在0.05水平,1-beta在0.20水平,根据Effect-Size计算出所需要的每组动物数量为8(G-Power3.0)。在本实验中,我们预定每组10只动物,以防动物意外死亡引起样本量的不足。根据脑缺血再灌注损伤的病理生理特点及我们前期的研究经验,设定在再灌注后8 h,以拉颈法处死动物。
1.4 大鼠NSS评分 采用双盲法,根据以下原则:(1)提鼠尾离地面约1尺,观察前肢情况。正常大鼠两前肢对称地伸向地面。有左肩内旋,左前肢内收者,评为4分,否则0分。(2)将动物置平滑地板上,分别推左(或右)肩向对侧移动,检查抵抗运动的阻力。正常大鼠两侧阻力明显对称。右肩向左侧移动时,发现阻力下降时,根据下降程度的不同,评为1~3分;(3)将动物两前肢置一金属网上,观察两前肢的张力,正常大鼠两前肢的张力明显对称,发现左前肢肌张力下降者,根据下降的轻重,评为0~3分。根据以上评分,满分10分,分数越高,说明动物的行为障碍越严重。
1.5 脑含水量测定 我们采用Hatashita的干湿重法测定脑含水量的变化。将完成NSS的实验动物处死,快速开颅取出大脑,并移到冰面,并测定大脑的湿重。再将动物大脑放入蒸箱中,在110 ℃,烘蒸24 h,测得大脑干重的量。含水率为(湿重大脑-干重大脑)/湿重大脑*100%。
1.6 免疫蛋白印记(WB)实验 将海马区域的皮质组织标本收集,并置入试管中,用冰冷的PBS洗涤两次;加入RIPA裂解缓冲液(Santa Cruze),含1%的蛋白酶抑制剂混合物。冰上匀浆、裂解10 min,低温离心12000 r/min,15 min,收集上清液,BCA法测定蛋白浓度。取30 μg总蛋白,经12%的SDS-PAGE Gel电泳分离,常规方法转印至硝酸纤维素(NC)膜上,5%脱脂牛奶-TBST封闭2 h,再分别将Cx40(1∶1000)、GAPDH(1∶2000)一抗加入封闭液,4摄氏度轻摇过夜,回收一抗液体,给予TBST溶液洗涤NC膜,加入辣根酶标记山羊抗兔二抗抗体与封闭液按1∶2000的比例混合液,37 ℃,孵育1 h,再采用化学发光试剂盒进行发光显影。采用Image J 1.36b软件下行吸光度分析,蛋白吸光度值/GAPDH为相对含量,以Sham组目标蛋白/GAPDH为100%,其余组与之进行比较。
1.7 ELFA检测氧化应激因子NAPDH氧化酶活性 自脑分离出海马的皮质组织,在冰上PBS中充分匀浆,以4 ℃,12000 r/min,10 min离心,取上清液,置入96孔荧光板中,再加入80 μlPBS及6.25 μl的1 mol/L的光泽精,再加入100 μmol/L的NAPDH后开始发生反应。采用多功能酶标仪,在吸收波长340 nm,检测速度30 s,时间为5 min。
1.8 ELISA法炎性因子IL-1β、IL-6和TNF-α的测定 采用第1.7部分的标本,按照炎性因子各自的ELISA免疫试剂盒的操作说明进行操作。
2 结 果
2.1 神经损伤严重度评分(Neurological Severity Score,NSS) GS-Rb1治疗组尽管较假手术组0分明显升高(P<0.05),但相对于溶媒组及损伤组却明显降低(P<0.05),具有治疗效应。采用H-89后,此种效应可被明显抑制(P<0.05)(见图1)。
2.2 脑含水量的测定 脑水肿含量测定结果表明,GS-Rb1治疗后,脑水肿程度较溶媒组及损伤组明显缓解(P<0.05),且与假手术组无明显差异(P>0.05)。但H-89可明显降低GS-Rb1的此种脑保护效应(P<0.05)(见图2)。
2.3 蛋白学检测Cx40在GS-Rb1及GS-Rb1+H-89单独及联合作用下表达的变化 在给予GS-Rb1治疗后,Cx40蛋白表达尽管较假手术组升高,但较溶媒组及损伤组明确下降,而H-89则可明显抑制GS-Rb1的此种效应(见图3)。
2.4 氧化应激因子NAPDH氧化酶(NAPDH oxidase activity)活性测定 GS-Rb1可明显下调损伤后神经元NAPDH氧化酶的活性(P<0.05),且与假手术组相比亦无明显差异(P>0.05),但是此种效应可被H-89明显的抑制(P<0.05)(见图4)。
2.5 ELISA检测海马区神经元氧化应激因子炎性因子IL-1β、IL-6及TNF-a等炎性因子蛋白的表达 GS-Rb1治疗组较阻断组可明显降低IL-1β、IL-6及TNF-a表达(P<0.05),但给予H-89后,则可明显抑制GS-Rb1的此种效应(P<0.05)(与假手术组比较*P<0.05;与GS-Rb1保护组比较#P<0.05)(见图5)。
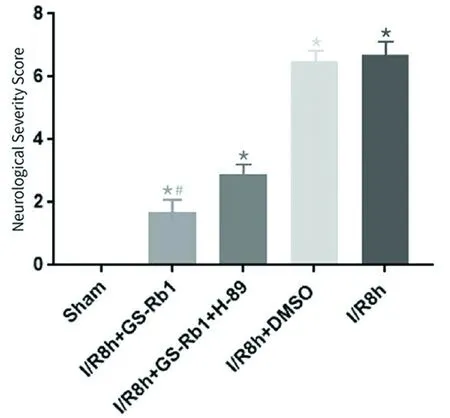
各实验组与假手术(Sham)组比较*P<0.05,I/R8 h+GS-Rb1组与/R8 h+GS-Rb1+H-89组比较#P<0.05
图1 NSS评分的变化(n=10)

各实验组与假手术(Sham)组比较*P<0.05,I/R8 h+GS-Rb1组与/R8 h+GS-Rb1+H-89组比较#P<0.05
图2 脑含水量的变化(n=10)

各实验组与假手术(Sham)组比较*P<0.05,# I/R8 h+GS-Rb1组与/R8 h+GS-Rb1+H-89组比较P<0.05
图3 Cx40相对含量的表达(n=10)

各实验组与假手术(Sham)组比较*P<0.05,# I/R8 h+GS-Rb1组与/R8 h+GS-Rb1+H-89组比较P<0.05
图4 NAPDH oxidase activity的变化(n=10)

A. IL-1β;B. IL-6;C. TNF-α各实验组与假手术(Sham)组比较*P<0.05,# I/R8 h+GS-Rb1组与/R8 h+GS-Rb1+H-89组比较P<0.05
图5 炎性因子的含量的变化(n=10)
3 讨 论
人参已经被广泛的作为药物、保健及饮食之用。人参皂甙(GS)已被证明是人参的最主要的活性成分。目前大量实验证明:GS具有抗炎、抗氧化及降低脂质类代谢产物的作用[4]。Rb1作为GS中含量最为主要的成分,得到越来越多的重视。目前已证实GS-Rb1能维持脑血屏障的完整,在脑缺血后治疗中起着明显脑保护作用。我们的前期研究发现:GS-Rb1可抑制氧化应激因子NOX1、NOX2、NOX4、NAPDH及炎症因子IL-1β,并上调细胞骨架蛋白ZO-1、Occludin的表达,具有神经功能保护作用[5]。但以上的研究尚未对GS-Rb1参与调节的损伤因子的具体方式和机制进行进一步分析,因此我们在本项实验中,我们采用信号传导通路的调节,来进一步观察GS-Rb1对上述产物的具体作用机制。
缝隙连接(Gap Junction,GJ)为细胞间的直接联系的方式。GJ可供细胞内的离子、某些小分子信号传导因子及细胞内容物(<1.2KD)在细胞间进行传递[6]。GJ在脑损伤的过程中起着重要的作用,不同类型的GJ对损伤/抗损伤信号具有选择性,但相关机制仍未完全清楚[7]。目前大量研究认为,Cx43和谷氨酰胺转移酶、Na-Ka-ATP酶、热休克蛋白相关性密切,可加强脑损伤性物质的转运,并减少氧化应激产物的产生[8,9],Cx43还可与Bcl2结合,缓解细胞凋亡的现象[10]。针对于Cx40蛋白与脑损伤的相关性,来自于动物模型及临床标本的研究均显示:脑损伤后Cx40蛋白含量的升高及Cx40与Cx43形成的异型缝隙连接的升高增多与脑损伤程度呈现一致性[11,12]。我们的前期研究还证实,Cx40的增多可能与氧化应激活性的增强密切相关[3]。此外,近年来的研究显示Cx40与IkBa具有同源性,其异常升高可能与核因子途径的激活相关[13]。因此,我们推测,Cx40蛋白的异常增多可能是Cx43功能的丧失的一种补偿机制,且异常表达Cx40对损伤/抗损伤物质的通过性平衡失调,导致损伤信号的扩大、氧化应激作用增强及核因子途径活化所致的炎性因子的大量增生,具有脑损伤的效应。同时在我们对上游的信号途径的筛选研究中发现,ERK1/2通路与Cx40异常增多及Cx43磷酸化的相关性最大,且通过抑制ERK1/2通路,可明显缓解脑损伤的程度[11]。有文献指出,在多条不同功能信号传导通路中存在竞争性抑制的Gatekeeper[14]效应,而PKA途径可上调Cx43的功能,从而被称为Cx43功能的Gatekeeper。因此我们推测,脑损伤后ERK1/2途径的激活可抑制PKA途径的活性,导致Cx40蛋白异常增多及Cx43蛋白磷酸化升高及缝隙连接功能紊乱可能与脑损伤密切相关。
结合上述的情况,我们认为GS-Rb1脑保护效应可能与PKA途径的激活、通过Gatekeeper效应抑制ERK1/2途径的活性从而进一步抑制Cx40升高有关。在我们的此项试验中,我们采用PKA的特异性抑制剂H-89,观察其如何影响GS-Rb1在脑损伤后的治疗效应,我们发现,H-89能明显抑制GS-Rb1下调Cx40的功能,同时能抑制GS-Rb1降低氧化应激因子及炎性因子等脑保护作用,并且这些作用均在形态学及行为学上得到了进一步的证实。
综上所述,我们的此项研究结果进一步明确GS-Rb1参与损伤后脑保护机制与抑制Cx40蛋白表达异常升高有着密切的关系,同时也明确了PKA途径可能参与了这个过程,从而以期为进一步研究脑缺血再灌注损伤可能的基因靶点治疗奠定基础。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
中国临床医学影像杂志(2022年5期)2022-07-26
昆明医科大学学报(2022年4期)2022-05-23
上海交通大学学报(医学版)(2022年3期)2022-05-05
昆明医科大学学报(2021年12期)2021-12-30
昆明医科大学学报(2021年2期)2021-03-29
中华养生保健(2020年9期)2021-01-18
中华养生保健(2020年4期)2020-11-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
癌变·畸变·突变(2015年3期)2015-02-27
