
果蝇不同细胞组分转录本多样性
2019-04-09 12:30:54刘秋芳
生物加工过程 2019年2期
刘秋芳,李 华
(上海交通大学生物医学工程学院,上海200240)
真核生物基因能够产生大量不同的mRNA产物,例如,在人类已注释的基因中,每个基因平均能够产生5个已知的转录本异构体,有些基因甚至能够产生多达80个转录本异构体(ensembl release 75)[1]。这些转录本异构体是由选择性转录起始、可变剪切以及多聚腺苷酸化等转录调控过程的共同作用产生的[2-4]。在动物、植物以及其他真核生物中,通过剪接的方式,将同一个基因的外显子按照多种组合方式拼接在一起形成许多不同的mRNAs[1]。mRNA的表达水平和对应蛋白的水平并不一致,目前,有很多科研工作人员致力于研究造成这种差异的原因[5-7]。研究人员在人类的研究中发现不同的可变剪切形式对蛋白的翻译水平有显著的影响,他们发现结合到多核糖体上的转录本异构体的3′UTR序列更短[8]。可变剪切对基因表达的影响目前比较公认的是通过2种方式进行的[8]:一是产生不同的可变剪切转录本异构体,从而增加蛋白的多样性;二是产生包含提前终止密码子的转录本,启动NMD(nonsense-mediated decay)通路降解该转录本,从而降低基因的表达水平。核糖体是蛋白质的加工场所,能够将mRNA序列通过特定的编码方式翻译成蛋白质。以前的研究主要着眼于基因水平上的mRNA丰度与蛋白质水平的关系,而忽视了转录本异构体本身与核糖体的结合在翻译中所起的作用。利用传统的核糖体图谱法可以收集并提取与核糖体结合的全长转录本,然后对这些与核糖体结合的转录本进行测序,可以得到翻译组数据[9]。通过对细胞质和核糖体结合RNA分别进行测序,可以得到这两种不同细胞组分的RNA。
本文中,笔者收集果蝇早期胚胎0~4 h的转录组(细胞质RNA)和翻译组(核糖体结合RNA)数据,并通过系统的比较分析来揭示果蝇不同细胞组分转录本多样性。
1 材料与方法
1.1 数据
本文中使用的果蝇早期胚胎0~4 h的转录组和翻译组数据来源于Li等[10]公布的数据。
1.2 方法
1.2.1 高通量数据分析
对于来自果蝇不同细胞组分的高通量测序数据,首先,使用TopHat 2(v2.0.9)分别将转录组和翻译组数据比对到果蝇的参考基因组上去[11]。接下来,使用Cufflinks(v2.2.1)的Cuffnorm模块,利用唯一比对的数据计算果蝇已知转录本的丰度,Cuffnorm计算转录本的表达水平时会将比对上参考基因组的reads数转化为FPKM值(fragments per kilobase of exon model per million mapped reads)[12]。最后,为了验证数据的可靠性,对来自不同细胞组分的数据进行相关性分析。
1.2.2 主要表达转录本分析
首先,为了得到不同细胞组分中主要表达的转录本,笔者分别对转录组和翻译组数据进行如下处理:一是删除低表达的基因,在某一细胞组分中,如果一个基因的任意一个转录本的表达值大于1,则在该细胞组分中保留该基因[13],否则删除该基因;二是计算不同细胞组分中主要表达的转录本;三是比较不同细胞组分中主要表达的转录本,找出主要表达转录本发生改变的基因。其次,对于不同细胞组分中主要表达转录本发生改变的基因,根据这些基因在不同细胞组分中主要表达转录本的非翻译区(UTR)和编码序列(CDS)是否发生变化进行分类。最后,将不同细胞组分中主要表达转录本CDS发生变化的基因用DAVID(v6.8)进行功能富集分析[14]。
2 结果与讨论
2.1 数据比对结果
为了得到不同细胞组分的转录本丰度,首先,使用TopHat 2(--library-type=fr-firststrand -G,其他为默认参数)分别将转录组和翻译组数据比对到果蝇的参考基因组上去。由于测序数据会比对到基因组的多个位置,这些比对结果可能是错误的比对,会对后续的分析造成一定的影响,因此,为了提高后续转录本丰度计算的准确性,去除掉多比对的数据,后续分析只使用唯一比对(unique mapping),数据结果见表1。由表1可以看到细胞质和核糖体结合RNA数据比对上的比率分别为94.49%和93.55%,唯一比对数据的比率分别为92.05%和90.55%,这说明所选择的数据具有较高的质量,能够很好地比对到参考基因组上去,可以用于后续的分析。

表1 比对前和比对后的数据
注:Cyto表示细胞质RNA数据(即转录组数据),Poly表示核糖体结合RNA数据(即翻译组数据)。
2.2 相关性分析结果
为了验证数据的可靠性,对早期胚胎细胞转录组和翻译组数据进行相关性分析。在相关性分析中,要求转录本的表达水平在2个细胞组分中的FPKM都高于1,以减少测序噪音对相关性的影响[13]。利用R语言中的cor.test函数计算转录组和翻译组数据的pearson相关系数,结果见图1。
由图1可知:早期胚胎细胞的转录组和翻译组的相关系数为0.85,这说明来自不同细胞组分的测序数据具有很好的相关性,符合预期,因此一定程度上支持了数据的可靠性。
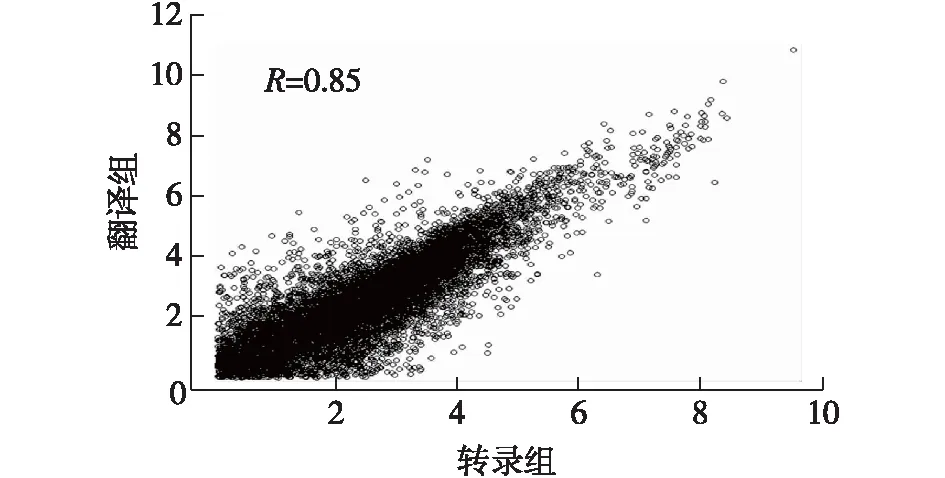
图1 果蝇0~4 h早期胚胎转录组和翻译组数据相关性Fig.1 Correlation between transcriptome data and translatome data of 0~4 h embryo cell in Drosophila melanogaster
2.3 不同细胞组分主要表达转录本分析结果
真核生物的基因表达水平与蛋白水平并不具有完全的一致性,有些基因通过选择性剪切能够转录得到多个转录本,每个转录本可能对应着不同的转录水平,不同的转录本可能具有不同的翻译效率。Floor等[1]研究发现,即使基因水平的表达丰度不变,在转录本水平上发生小小的改变也会对蛋白的水平产生巨大的影响。核糖体是转录本翻译成蛋白质的场所,因此,笔者想要研究是否是细胞质和核糖体结合RNA中转录本组成差异造成基因与蛋白表达水平的不一致性。
果蝇已知基因数是17 746个,已知转录本数是35 113个,平均每个基因含有2个转录本。为了得到不同细胞组分中主要表达的转录本,首先分别对果蝇转录组和翻译组数据的基因进行筛选,结果见图2。
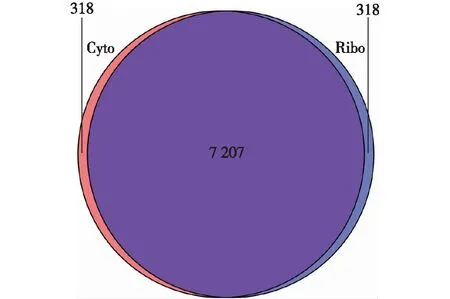
图2 果蝇早期胚胎不同细胞组分表达的基因重合数Fig.2 Overlap of expressed gene number of different cellular compartments in Drosophila melanogaster
由图2可知:在转录组和翻译组数据中剩余的基因数分别是7 525个(占所有已知基因的42.4%)和7 525个(占所有已知基因的42.4%),不同细胞组分的基因重合数是7 207个(其中表达2个及2个以上转录本的基因数为3 650个,占重合基因总数的50.65%),表明不同细胞组分中表达的基因具有较高的一致性。然后,通过计算和比较分析,在表达2个及2个以上转录本的重合基因中找出了主要表达的转录本发生变化的基因数766个。接下来,对这些基因按照不同细胞组分中主要表达的转录本UTR和CDS是否发生变化进行分类,UTR和CDS的信息来源于Flybase数据库发布的r6.13版本的果蝇已知注释信息(ftp://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.13_FB2016_05/gtf/)[15]。
经分类计算,发现400个基因的UTR发生变化,366个基因的CDS发生变化。最后对CDS发生变化的基因用DAVID进行GO功能富集分析和KEGG通路分析,结果见表2~3。由表2可知,这些基因与蛋白结合(protein binding)、GTP酶激活活性(GTPase activator activity)等分子功能相关。由表3可知,这些基因参与到果蝇早期胚胎背腹轴的形成过程。
以上结果表明,基因在细胞质和核糖体结合RNA中主要表达的转录本发生了改变。核糖体是蛋白质的生产场所,这可以在一定程度上解释蛋白质表达水平和转录本表达水平的不一致性。
2.4 讨论
笔者收集了果蝇早期胚胎细胞的转录组和翻译组数据,并通过系统的分析展示了不同细胞组分中RNA的组成差异。具体而言,首先利用TopHat 2软件将转录组和翻译组数据比对到果蝇的参考基因组上,得到不同细胞组分中的转录本表达丰度。然后,对于表达多个转录本的基因,在细胞质和核糖体结合RNA数据中分别找到其主要表达的转录本。最后,对于在2个样本中都表达的基因,计算了这些基因主要表达转录本的组成异同。最终,找到了766个主要表达转录本发生差异的基因,这表明在果蝇中大量基因的不同转录本的翻译效率差异可能很大。通过对基因序列特征的分析,将主要表达转录本发生变化的基因分为两类:UTR变化类(400个基因)和CDS变化类(366个基因)。转录本CDS的改变意味着编码得到的蛋白也发生了改变,这可以一定程度上解释蛋白质表达水平和转录本表达水平的不一致性。
由于果蝇的基因与人类的基因存在比较大的同源性,对果蝇胚胎发育的研究成果可以一定程度地应用在人类身上,为了解人类的胚胎发育过程提供一定的依据,因此,对果蝇不同细胞组分转录本多样性的研究是很有价值的。

表2 果蝇早期胚胎细胞中CDS发生变化的基因功能富集分析结果

表3 果蝇早期胚胎细胞中CDS发生变化的基因KEGG通路分析结果
3 结论
笔者对果蝇不同细胞组分转录本组成多样性进行了初步的研究,对CDS发生变化的基因进行功能富集分析和KEGG通路分析,发现这些基因显著富集到某些分子功能上并在特殊的通路中发挥功能。另外,UTR是序列上的非编码区域,对转录本的翻译起着重要的调控作用。因此,将来有必要对UTR上的调控元件做进一步的研究。
(责任编辑 荀志金)
猜你喜欢
学苑创造·A版(2023年10期)2023-11-04 13:14:04
大自然探索(2023年11期)2023-03-01 09:04:36
内蒙古民族大学学报(自然科学版)(2022年2期)2022-11-22 06:44:43
学苑创造·A版(2022年3期)2022-03-29 23:32:16
中国生殖健康(2020年5期)2021-01-18 02:59:48
肿瘤防治研究(2020年5期)2020-07-09 13:06:08
生物学教学(2019年9期)2019-09-23 03:53:02
学苑创造·A版(2019年6期)2019-07-11 01:07:39
中国生殖健康(2018年5期)2018-11-06 07:15:38
新疆医科大学学报(2015年10期)2015-12-26 12:33:30
