
无溶剂法原位合成多级孔Fe@MFI催化剂对费托合成反应性能的影响
2019-04-08 01:42程世林张桂华石嫣雯郭凯梁吕成学
石油化工 2019年3期
程世林,张桂华,石嫣雯,郭凯梁,吕成学,2,邢 闯,2
(1. 浙江科技学院 生物与化学工程学院,浙江 杭州 310023;2. 浙江科技学院 浙江省农产品化学与生物加工技术重点实验室,浙江 杭州 310023)
合成气(CO+H2)催化转化制碳氢化合物的费 托合成反应被认为是非油基碳资源转化利用的重要技术[1]。虽然费托合成技术日趋成熟,但依然存在着诸多技术难题,如汽油馏分C5~11选择性最高约为45%,难以定向合成目标产物[2]。因此,设计和制备高性能费托催化剂,实现目标产物选择性的有效调控,是亟待解决的关键性问题之一。
近年来,将分子筛引入费托合成,构建Metal@Zeolite核壳或封装型催化剂,在调节产物对汽油烃类选择性方面有显著的效果[3-5]。Sun等[6]将铁铝合金与强碱性的HZSM-5沸石合成液水热晶化,晶化过程中Al被脱除,一步制备了骨架Fe封装在HZSM-5沸石内部的核壳催化剂R-Fe@HZSM-5;Javed等[7]采用水热法在Co/HZSM-5分子筛上生长了一层Silicalite-1沸石壳,Co粒子被完全封装在夹层内。这些封装型催化剂由于具有空间受限、独特的孔道和较高的扩散效率等特点在费托催化汽油馏分异构化反应中显示出良好的应用前景。本教研组借鉴Ren等[8]提出的无溶剂合成沸石路线成功制备出多种Co基分子筛催化剂,并运用于费托反应中,取得了较高的汽油收率[9-10]。
本工作联合浸渍法-无溶剂法制备了一系列负载型和封装型催化剂,通过XRD、N2吸附-脱附、NH3-TPD、SEM和TEM等手段对催化剂进行物化性能表征,并考察了所制备催化剂对合成气直接转化成汽油烃类的反应性能的影响。
1 实验部分
1.1 催化剂的制备
1.1.1 负载型Fe/SiO2催化剂的制备
等体积浸渍法制备Fe/SiO2和Fe/HZSM-5催化剂。采用Fe(NO3)3·9H2O水溶液在两种不同孔径(10 nm和50 nm)SiO2载体上负载10%(w)的Fe,然后进行真空干燥1 h,400 ℃空气煅烧2 h,制得催化剂记为 10Fe/SiO2(10)和 10Fe/SiO2(50)。
Fe/HZSM-5的制备过程同上。
1.1.2 封装型Fe@MFI催化剂的制备
采用无溶剂原位合成路线,“一步法”制备多级孔Fe@MFI沸石催化剂。将一定量的NaOH、勃姆石、Fe/SiO2及乙二胺于研钵中充分研磨混合30 min后,再转移至反应釜中,在恒温箱内200 ℃下晶化72 h。将晶化好的前体进行过滤、干燥、焙烧,制得的催化剂,记为Fe@NaZSM-5(10)和Fe@NaZSM-5(50)。将2 g Fe@NaZSM-5与30 mL的2 mol/L的NH4NO3溶液置于100 mL的圆底烧瓶中,在80 ℃水浴锅内离子交换4 h,然后进行过滤、干燥、煅烧,制得催化剂,记为Fe@HZSM-5(10)和Fe@HZSM-5(50)。
1.2 催化剂的活性测试
费托反应活性评价在自制高压固定床反应装置(6.8 mm×550 mm)上进行,催化剂位于固定床反应器等温区内,催化剂床层由混合均匀的20~40目0.3 g催化剂和0.6 g石英砂组成,上端和下端分别使用石英棉固定。反应前先在合成气(V(H2)∶V(CO)∶V(Ar)=64∶32∶4)氛围中在线还原活化4 h,还原条件为常压,300 ℃,流速40 mL/min,升温速率2 ℃/min。还原结束后,保持温度不变,系统压力调至1.0 MPa,流速降为20 mL/min。当系统压力、温度和流速稳定后,开始在线进样。分别使用带有TCD和FID的上海天美公司GC7900型气相色谱对原料和产物进行在线分析;反应结束后,对冷阱中的液相产物用带有FID的岛津公司GC-2014型气相色谱进行离线分析,并计算反应物转化率和产物选择性。
1.3 催化剂的表征
采用日本理学公司Rigaku Ultima Ⅳ型X射线衍射仪进行催化剂的物相分析,利用Scherrer公式计算金属物种的晶粒大小。采用美国康塔公司的Autosorb-iQ型全自动物理化学吸附仪进行催化剂的比表面积和孔结构分析,采用静态法测定试样的N2吸附-脱附等温线。在日本麦奇克拜耳公司BELCAT-B3型化学吸附仪上进行NH3-TPD 测试,首先试样在300 ℃下用流速为25 mL/min的He吹扫1 h,然后在30 mL/min的 5%(φ)NH3/He混合气吹扫下,以10 ℃/min的速率升温至800 ℃,采用TCD检测NH3的脱附量。在美国FEI公司Quanta 400 FEG 型扫描电子显微镜下对试样的形貌进行观察,测试前将粉末试样进行真空干燥,喷金处理。利用美国FEI公司Tecnai G2 F20 型场发射电子显微镜对试样内部结构进行观察,加速电压为200 kV。
2 结果与讨论
2.1 催化剂的XRD分析结果
图1为反应前催化剂的XRD谱图。由图1可知,封装型催化剂Fe@MFI的XRD谱图具有明显MFI骨架结构的五指峰,且无其他杂峰。负载型催化剂 Fe/SiO2和 Fe/HZSM-5在 2θ=33.1°,35.6°,40.8°,49.5°,54.1°,62.5°,64.0° 处均出现了α-Fe2O3的特征衍射峰。Fe@MFI催化剂只在2θ=35.6°(110)处观察到α-Fe2O3的特征衍射峰,而其他衍射角没有明显的衍射峰,这可能是由于原位合成Fe@MFI过程中Fe更容易暴露出较多的(110)晶面,其他晶面可能受到分子筛的遮蔽,从而未能达到XRD的检测限。此外,相比负载型催化剂,封装型Fe@MFI催化剂在2θ=35.6°处的衍射峰峰形变宽,说明Fe晶粒更小。与Na型Fe@NaZSM-5相比,H型Fe@HZSM-5晶型并未发生明显变化,说明离子交换和二次煅烧不会对催化剂的晶型产生影响。
2.2 催化剂的比表面积和孔结构分析结果
图2为不同催化剂的N2吸附-脱附曲线。由图2a可知,催化剂Fe/HZSM-5的N2吸附曲线属于Ⅰ类吸附等温线,是典型的微孔材料。4种Fe@MFI催化剂属于Ⅳ类吸附等温线并伴有明显的H4型滞后环,出现这一现象是由于介孔的引入造成。同时,带有H4型滞后环的吸附分支是由Ⅰ型和Ⅱ型等温线复合而成,在相对压力较低端有非常明显的吸附量,说明存在微孔填充。另外,可能是离子交换作用使得H+占据了被交换离开的Na+空位,由于H+半径明显小于Na+,导致微孔体积的增加,呈现出H型催化剂Fe@HZSM-5在相对压力较低端吸附量增加,滞后环明显缩小的现象。由图2b可知,Fe/SiO2(10)和Fe/SiO2(50)的N2吸附等温曲线呈Ⅳ型,属于介孔材料。

图1 反应前催化剂的XRD谱图Fig.1 XRD patterns of the calcined catalysts.

图2 不同催化剂的N2吸附-脱附曲线Fig.2 N2 adsorption-desorption curves of synthesized catalysts.
图3为负载型和封装型催化剂的孔径分布。由图3可知,催化剂Fe/HZSM-5和两种Na型Fe@NaZSM-5的最可几孔径为0.65 nm,平均孔径大约在0.56~0.9 nm左右。而经离子交换后的H型催化剂Fe@HZSM-5的最可几孔径为0.54 nm,往小孔径的地方发生了迁移,缩减了大约0.11 nm;同时,最可几孔径对应的孔体积扩大了约3.5倍,平均孔径的范围也大大缩小,在0.48~0.6 nm。这些现象与图2a中H型催化剂Fe@HZSM-5的吸附等温线在低压端吸附量增加,导致微孔体积增大的情况一致。表明通过离子交换作用清理了堵塞在沸石原有孔道中的无定型物种和Na+,沸石的微孔孔道得以疏通,从而暴露出更多的微孔孔道。
催化剂的物性参数见表1。从表1可知,与两种Fe/SiO2催化剂相比,两种Na型Fe@NaZSM-5的α-Fe2O3晶体粒径分别缩减9.9 nm和4 nm,这是由于在原位合成Fe@MFI的过程中,可能受到了NaOH的溶解作用,导致了α-Fe2O3晶体粒径大大减小。由文献[11-12]可知,NaOH的加入不仅可以平衡分子筛Si/Al骨架电荷,还会发生部分碱性溶Si过程,碱处理是产生晶内介孔的主要原因,产生的多级孔道结构可以有效改善催化剂的传质扩散性能。H型Fe@HZSM-5比Na型Fe@NaZSM-5表现出更大的比表面积和微孔体积,说明离子交换作用对催化剂的比表面积和孔结构有很大影响。另外,离子交换作用带走了部分覆盖在α-Fe2O3晶粒上的Na+导致H型Fe@HZSM-5的α-Fe2O3晶体粒径尺寸有所减小。

图3 负载型和封装型催化剂的孔径分布Fig.3 Pore size distribution patterns of supported and encapsulated catalysts.

表1 催化剂的物性参数Table 1 The physical parameters of synthesized catalysts
2.3 催化剂酸性分析结果
NH3-TPD 的峰面积和出峰温度可反映催化剂的酸量和酸强度。不同催化剂的NH3-TPD谱图见图4。由图4可知,无定形SiO2作为载体不显酸性,HZSM-5分子筛作为载体呈现出2个NH3脱附峰,封装型Fe@MFI催化剂均出现3个NH3脱附峰。120~250 ℃段低温NH3脱附峰对应弱酸中心,250~350 ℃段中高温NH3脱附峰对应中等强酸中心,350~500 ℃段高温NH3脱附峰对应强酸中心。观察峰面积可知,相比Na型Fe@NaZSM-5,H型Fe@HZSM-5在中高温段NH3脱附峰面积大大减小,低温段和高温段NH3脱附峰面积有所增加,导致对应的中强酸显著减少,弱酸和强酸少量增加。说明在制备Na型Fe@NaZSM-5过程中,Na+进入阳离子交换位,会导致催化剂中B酸酸性显著减弱;通过将Fe@NaZSM-5中的Na+与NH4+交换制得H型Fe@HZSM-5,可部分恢复强B酸酸性。由此可见,通过离子交换改变Na+的含量来调控酸性位和酸中心数量是可行的。

图4 不同催化剂的NH3-TPD谱图Fig.4 NH3-TPD patterns for all the different catalysts.
2.4 催化剂的形貌分析结果
图 5为 Fe/SiO2(10)和 Fe@NaZSM-5(10)催化剂的SEM和TEM照片。由图5a可看出,铁粒子团聚现象明显,说明负载型催化剂的活性组分粒子大小和分散性难以控制,易形成团簇。图5b和5c是将Fe/SiO2(10)作为Si源原位合成Fe@NaZSM-5(10)的TEM和SEM照片,这种原位合成技术制备出的微米级内嵌Fe粒子的ZSM-5沸石,形貌均一,呈六方形结构,但棱角不太明显,存在较严重的团聚现象。这是因为在无溶剂结晶过程中,存在着非流动配位体系,使得部分非晶相组分未能完全转化而附着在沸石单晶上,使之呈现不太完美的ZSM-5形貌,这些非晶相组分主要来源于未转化的固体原料。图5d 中ZSM-5沸石的晶格条纹清晰可见,孔径多集中在2~3 nm之间,部分Fe粒子非常均匀地分散在沸石介孔中。
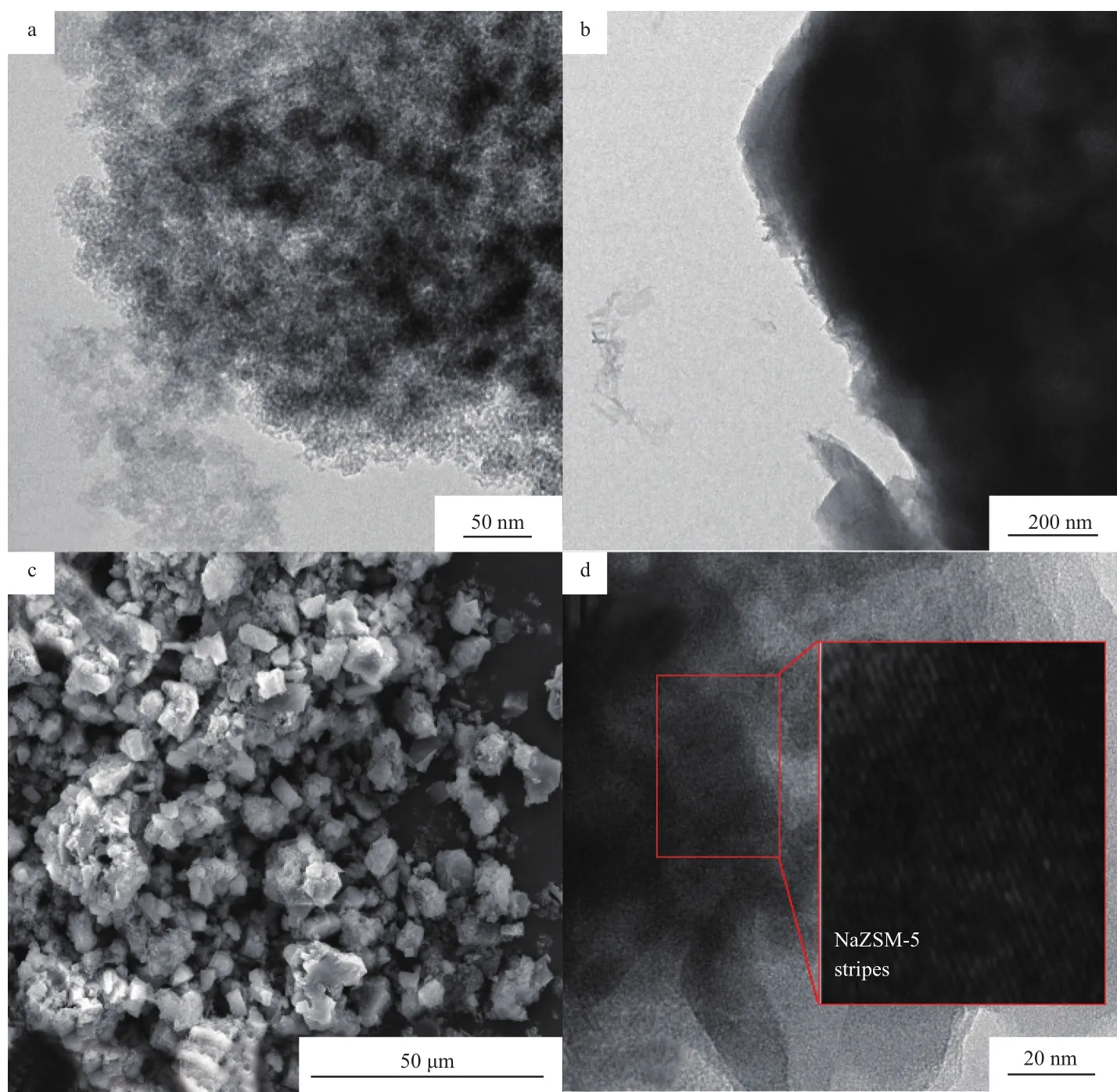
图5 Fe/SiO2(10)和Fe@NaZSM-5(10)催化剂的TEM(a,b,d)和SEM(c)照片Fig.5 TEM(a, b, d) and SEM(c) images of synthesized Fe catalysts.
2.5 催化剂的费托合成反应性能
在反应温度300 ℃ 、压力1 MPa、相对质量流速为6(g·h)/mol的条件下,对负载型和封装型催化剂进行了费托反应性能评价,结果见表2。
负载型和封装型催化剂显示出较大的CO加氢反应活性差异,归因于它们的制备方法不同使结构特征和理化性质发生了变化。从表2可知,负载型催化剂由于孔道结构和载体酸性等因素展现出不太好的产物分布。Na型Fe@NaZSM-5具有较多的中强酸性位和更加宽化的微孔孔道(0.56~0.9 nm),会促进小分子的低碳烃和大分子的重质烃生成,不利于目标产物的定向合成。H型Fe@HZSM-5具有较低的CH4,CO2选择性及更高的C5~11选择性,说明通过离子交换减少Na+含量使更多的金属活性中心和弱酸中心暴露,可以有效地抑制CH4的生成和降低水煤气反应活性,从而提升低碳烃的加氢反应,有利于短链烃向中长链烃转化。与Fe@HZSM-5(50)相比,Fe@HZSM-5(10)的C5~11选择性达到了58.9%,CO2和CH4选择性分别降为23.8%和12.6%。这是因为,Fe@HZSM-5(10)具有更高比表面积,有利于合成气和产物的吸附,较多适合汽油馏分产生的微孔(0.48~0.6 nm)孔道以及较丰富的弱酸中心有利于正构烷烃的异构化反应。

表2 催化剂的费托合成性能Table 2 The catalytic performance of synthesized catalysts in Fischer-Tropsch synthesis
图6为催化剂的汽油和异构烷烃选择性。由图6可知,负载型和Na型Fe@NaZSM-5催化剂均表现出低于50%的汽油馏分和低于30%的异构烷烃选择性;两种H型Fe@HZSM-5表现出较高的汽油馏分和异构烷烃选择性,造成这种差异的原因主要归咎于它们的孔结构和表面酸性不同。通常,沸石酸度和孔道结构是影响异构烷烃选择性的主要因素[13-14]。将Fe@NaZSM-5经过离子交换作用转化成酸度适宜的H型Fe@HZSM-5沸石,比表面积、孔结构及酸性得到了改善,从而表现出更高的汽油馏分和异构烷烃选择性。同时由于H型沸石中占优势的弱酸性有利于碳正离子的去质子化反应,降低了进一步发生裂化反应的概率,使得异构化反应的选择性提高,从而避免了过多裂化反应的发生[15]。
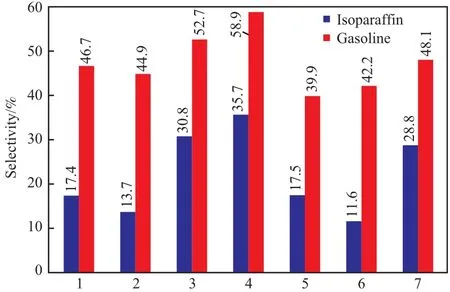
图6 汽油和异构烷烃的选择性Fig.6 Gasoline and isoparaffin selectivities of the prepared catalysts.
3 结论
1)采用无溶剂法成功合成了具有MFI骨架结构的Fe@MFI催化剂,Fe粒子较好地封装在沸石内部;催化剂的孔结构和酸中心的协调作用是影响费托合成产物分布的主要因素。
2)与负载型Fe/SiO2催化剂相比,封装型Fe@MFI催化剂在费托合成反应中表现出良好的汽油选择性,该催化剂的多级孔结构与酸中心的协同效应发挥了作用。
3)离子交换作用有效改善了H型Fe@HZSM-5催化剂的孔结构和表面酸性,使得汽油和异构烷烃的选择性得到了明显提升。
猜你喜欢
内燃机与动力装置(2022年1期)2022-03-21
煤气与热力(2021年9期)2021-11-06
化学工业与工程(2021年5期)2021-11-03
湖南饲料(2021年3期)2021-07-28
湖北农机化(2020年4期)2020-07-24
天然气化工—C1化学与化工(2019年6期)2019-02-18
筑路机械与施工机械化(2017年5期)2017-08-31
天津大学学报(自然科学与工程技术版)(2015年10期)2015-12-29
