
盐藻钙依赖蛋白激酶基因DsCDPK的表达分析
2019-03-29 07:14丛玉婷邢震宇岳金荣高相楠张晓琳柴晓杰
水产科学 2019年2期
丛玉婷,邢震宇,岳金荣,高相楠,张晓琳,柴晓杰
( 大连海洋大学,辽宁省省级高校水生生物学重点实验室,辽宁 大连 116023 )
盐藻(Dunaliellasalina)是己知最为耐盐的单细胞真核生物,它能在低盐和高盐(NaCl的浓度为0.05~5.0 mol/L)的极端环境中生存,已引起了生物学家的广泛关注,并成为耐盐植物中的模式生物。一直以来,国内外学者从渗透调节[1-3]、耐盐基因[4-5]和盐适应蛋白质[6-7]等多个方面对其进行了深入研究,并取得了一些研究成果。但盐藻应答盐胁迫信号的分子机制目前尚不清楚。
钙依赖蛋白激酶(CDPK)是一种新型的仅仅依赖Ca2+而不依赖钙调素的蛋白激酶。当结合上Ca2+后该蛋白才具有活性,活化状态的蛋白分子具有丝氨酸/苏氨酸蛋白激酶的活性[8]。钙依赖蛋白激酶广泛存在于植物、藻类及部分原生动物中,尚未在细菌、真菌、酵母、线虫和动物中发现[9]。研究表明,钙依赖蛋白激酶广泛参与了Ca2+信号转导通路介导的细胞碳氮代谢[10]、逆境胁迫应答[11]、植物细胞膜系统的物质运输[12-13]、植物细胞肌动蛋白张力的调节[14-15]和生长发育调节[16]等诸多生物学过程,特别是在抗逆胁迫信号转导途径中起到了关键作用。因此,研究盐藻钙依赖蛋白激酶基因的功能,对于阐明盐藻耐盐的分子机制和信号转导途径具有重要的科学意义。
笔者自盐藻中提取其总RNA,以总RNA为模板,利用实时荧光定量PCR技术获得盐藻钙依赖蛋白激酶基因DsCDPK,构建原核表达载体并在大肠杆菌(Escherichiacoli)中成功表达;通过实时荧光定量PCR技术分析在盐胁迫条件下DsCDPK基因的表达情况,以期为进一步阐明盐藻适应高盐环境的分子机制及盐藻应答盐胁迫信号的分子途径提供参考。
1 材料与方法
1.1 材料
盐藻藻种由大连海洋大学水生生物学实验室提供。盐藻培养液为康威试液[17],25 ℃,12 h光照(光照度为1250 lx),12 h黑暗,进行交替静置培养。大肠杆菌BL21,pET-32a由本室保存,pMDTM19-T simple购自TaKaRa公司。
RNAiso Plus、DNA Marker DL-2000、限制性内切酶、Taq酶、溶菌酶、IPTG、X-gal、DEPC、Reverse Transcriptase M-MLV (RNase H-)均购自TaKaRa公司,凝胶回收试剂盒购自爱思进生物技术有限公司,其他分析纯试剂均为科密欧生产厂家提供。
1.2 盐藻总RNA的提取
取对数生长期的盐藻10 mL(培养液含3.0 mol/L NaCl),离心收集藻细胞,按照RNAiso Plus试剂盒(TaKaRa, China)操作说明书提取总RNA。经1%琼脂糖凝胶电泳检测RNA含量。
1.3 盐藻DsCDPK基因的克隆及原核表达载体的构建
根据DsCDPK基因cDNA全长序列(GenBank: JQ964113)设计引物,N1:5′-CGGAATTCATGGGGTGTTCGGACAGCAAACCTG-3′(下划线处为EcoRⅠ酶切位点)和N2:5′-GTCGACTCAAGCCGTGGCCTTGACCATGGGT -3′(下划线处为SalⅠ酶切位点)。用总RNA反转录成cDNA,以cDNA为模板,N1、N2为引物进行PCR扩增。PCR反应体系:94 ℃预变性5 min;94 ℃变性30 s,60 ℃退火30 s,72 ℃延伸2 min,35个循环;72 ℃延伸10 min。目的片段与pMDTM19-T simple载体连接,获得阳性单克隆。将重组质粒pMDTM19-DsCDPK和pET-32a质粒均用EcoRⅠ、SalⅠ双酶切,回收目的片段及载体大片段,T4 DNA 连接酶连接,重组克隆经筛选、鉴定及测序。
1.4 盐藻DsCDPK基因的原核表达及融合蛋白的可溶性分析
将测序正确的pET-32a-DsCDPK重组质粒转化到大肠杆菌BL21(TIANGEN公司)感受态细胞,挑取单菌落,接种于5 mL LB液体培养基(Amp 50 mg/L)中,37 ℃振荡培养(200 r/min)24 h。取1 mL菌液转接到50 mL LB液体培养基(Amp 50 mg/L)中,37 ℃,200 r/min培养至OD600为0.6~0.8,加入IPTG至终浓度为1 mmol/L,培养5 h后(同时设置对照)离心收集沉淀,再加入0.1 mol/L PBS 10 mL重悬,在冰浴中用超声波细胞破碎仪进行破碎细胞20 min(功率30 W,工作9 s,间歇9 s)。8000 r/min离心5 min,分别收集上清液和沉淀,进行SDS-PAGE分析。
1.5 重组蛋白的Western杂交检测
用超声波细胞破碎仪破碎诱导后的菌液(冰浴,间歇时间为10 s,10 min),12 000 r/min,4 ℃离心20 min后取上清液用0.45 μm滤膜抽滤。然后用His60镍柱纯化试剂盒(Clontech公司)纯化,纯化产物经SDS-PAGE分离,电转移(200 mA,3 h)至硝酸纤维素膜,用3% BSA 4 ℃封闭24 h,然后将硝酸纤维素膜置于用0.01 mol/L 磷酸盐缓冲液按1∶1000比例稀释的Anti-His Antibody(TIANGEN公司)中,37 ℃孵育1 h,用0.01 mmol/L PBST洗涤3次,每次5 min。然后加入二抗(TIANGEN公司)HRP-IgG (1∶200),37 ℃孵育1 h,洗涤(方法同上),最后使用TMB显色液(TIANGEN公司)进行显色。
1.6 盐胁迫下盐藻DsCDPK基因的表达分析
盐藻在培养液中生长至对数生长期(培养液含1.0 mol/L NaCl),此时加入NaCl致使其盐浓度达到3 mol/L。分别提取胁迫0(对照)、0.5、1、6、12、24 h的盐藻的总RNA。用紫外分光光度法检测RNA样品的纯度和含量,然后取各RNA样品500 ng反转录。目的基因引物为D1:5′-GGACTTTGGGCTGTCTCGTTT-3′和D2: 5′-CTTGGCTGCGTCTGTGATCTT-3′;18S内参基因引物为N1: 5′-TTGGGTAGTCGGGCTGGTC-3′,N2: 5′-CGCTGCGTTCTTCATCGTT-3′。荧光定量PCR反应在ABI 7300 Real-Time PCR System上进行,反应体系为:SYBR Premix Ex TaqTMⅡ (TaKaRa, China)10 μL,上下游引物(10 μmol/L)各0.8 μL,ROX Reference Dye (50×) 0.4 μL,cDNA模板2 μL,ddH2O 6 μL。反应条件为:95 ℃预变性30 s;95 ℃变性5 s,60 ℃退火34 s,40个循环,反应结束后确认扩增曲线和融解曲线。采取2-ΔΔCt法计算DsRab基因的相对表达量[20]。采用SPSS软件进行数据统计分析。
2 结果与分析
2.1 盐藻DsCDPK基因的克隆及原核表达载体的构建
以盐藻总RNA为模板进行PCR扩增,用1%琼脂糖凝胶电泳分析PCR产物(图1a)。在约2000 bp位置有一条清晰的条带,与试验预期结果一致。重组质粒pMD19-T Simpie-DsCDPK经双酶切及质粒PCR检测,结果表明,目的片段已连接到克隆载体pMD19-T simple上(图1b)。重组质粒的测序结果与目的基因序列比对后,与DsCDPK开放阅读框完全一致,表明重组质粒pMD19-T Simpie-DsCDPK构建成功。将已经鉴定正确的克隆载体pMD19-T Simpie-DsCDPK和原核表达载体pET-32a进行EcoRⅠ、SalⅠ双酶切,经T4连接酶连接,构建原核表达载体pET-32a-DsCDPK,转化大肠杆菌BL21感受态,筛选出阳性单克隆。经EcoRⅠ、SalⅠ双酶切和质粒PCR检测(图1c),结果显示,目的片段大小与预期一致,说明目的片段与原核表达载体正确连接。测序结果表明,扩增片段为1650 bp,与DsCDPK(GenBank: JQ964113)开放阅读框完全一致,读码框正确,原核表达载体pET-32a-DsCDPK构建成功。

图1 盐藻DsCDPK的PCR及酶切产物的琼脂糖凝胶电泳分析a.M: DNA marker DL-2000,1: PCR 产物;b.M: DNA marker DL-5000,1: 酶切产物,2: PCR产物;c.M: DNA marker DL-15 000,1: 重组质粒,2: 酶切产物,3: PCR产物.
2.2 盐藻DsCDPK基因的原核表达及重组蛋白的Western杂交检测
将pET-32a-DsCDPK及pET-32a转化至表达菌大肠杆菌BL21感受态细胞中,含有空载体pET-32a的表达菌为对照组,含有pET-32a-DsCDPK重组质粒的表达菌,经IPTG诱导后,电泳结果显示,约在82 ku处有一条明显增强的条带,与预期结果相符(融合蛋白包括预计分子量为61 ku的DsCDPK蛋白和21 ku的His标签蛋白),而含有空载体pET-32a的表达菌,未显示该目的条带,表明DsCDPK基因在大肠杆菌中成功表达。诱导后的大肠杆菌表达菌经超声波破碎,取上清液和沉淀分别电泳,结果表明,上清液和沉淀均可见清晰的目的蛋白条带,说明目的蛋白为部分可溶性表达(图2)。可溶性蛋白经His60镍柱纯化试剂盒纯化,纯化产物经Western blotting检测,在82 ku处有明显的单一蛋白条带(图3),说明融合蛋白能与抗His单克隆抗体特异性结合,具有良好的免疫学活性,初步证明纯化的蛋白就是带有His标签的钙依赖蛋白激酶。

图2 融合蛋白的诱导表达和表达形式分析M:标准蛋白分子量; 1:含pET-32a质粒的表达菌株经IPTG诱导; 2:含pET-32a-DsCDPK 的表达菌株经IPTG诱导后在上清液中的表达; 3:含pET-32a-DsCDPK的表达菌株经IPTG诱导后在沉淀中的表达.

图3 Western blotting检测M:标准蛋白分子量,1:纯化的融合蛋白.
2.3 盐胁迫条件下DsCDPK基因的表达分析
通过实时荧光定量PCR方法分析了盐藻DsCDPK基因在盐胁迫条件下的表达模式,结果见图3。在0 h正常生长条件下(未胁迫),DsCDPK基因的表达量很低,当受到3.0 mol/L NaCl胁迫时,DsCDPK基因的表达量明显升高,在胁迫1 h时表达量是未胁迫时的3倍,并且达到最高,差异达极显著水平(P<0.01)。随着胁迫时间的延长,表达量有所下降,但仍高于正常情况下的表达量(图4),表明DsCDPK为盐胁迫上调基因,可能在盐藻应答高盐胁迫的生理过程中起重要作用。
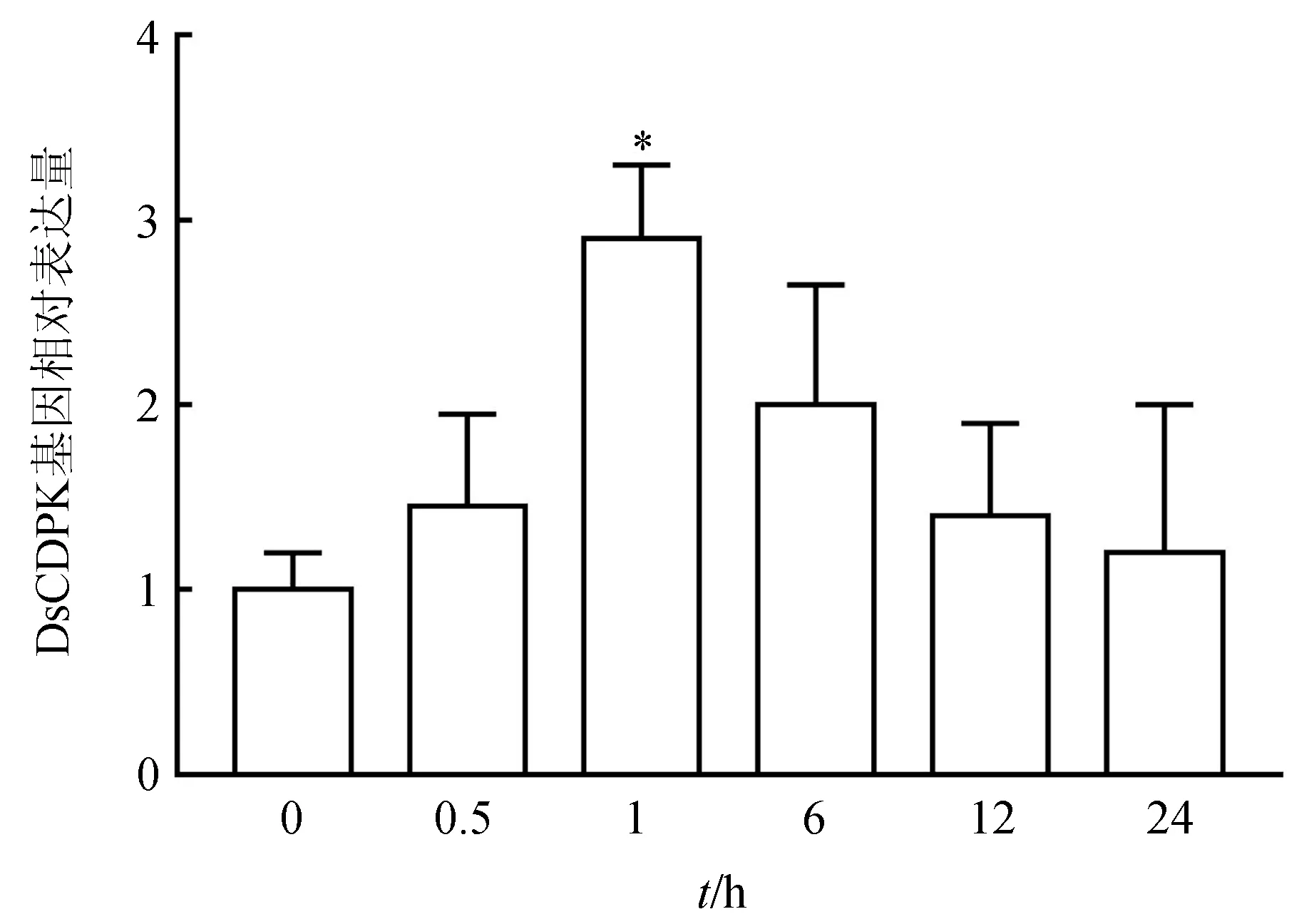
图4 盐藻DsCDPK 基因在不同盐胁迫时间下的表达分析
3 讨 论
3.1 钙依赖蛋白激酶参与植物应答盐胁迫过程的调控
细胞在各种外界信号刺激下做出的应答反应主要是通过蛋白激酶对通路蛋白的磷酸化修饰、蛋白质的相互作用以及下游靶基因表达的改变等一系列生化事件最后导致特定的生物效应。研究表明,钙依赖蛋白激酶广泛参与了逆境胁迫应答和生长发育等多种Ca2+信号传递途径中信号的介导。盐胁迫、干旱胁迫、冷胁迫、光照及渗透胁迫等多种逆境胁迫,均能诱导钙依赖蛋白激酶基因的特异性表达并引起钙依赖蛋白激酶在 mRNA水平的特异性积累。在拟南芥中发现的钙依赖蛋白激酶基因cATCDPK1和cATCDPK2,在高盐和干旱胁迫下其mRNA水平的表达被迅速诱导[11]。Saijo等[18]在转基因水稻中对钙依赖蛋白激酶基因进行了盐胁迫、干旱胁迫和冷胁迫的表达分析,研究发现,水稻应对盐胁迫(200 mmol/L NaCl)时OsCDPK7基因的表达量增加。Kiselev等[19]研究发现,人参钙依赖蛋白激酶基因PgCDPK1c、PgCDPK2c和PgCDPK4a在60 mmol/L NaCl盐浓度下表达量明显增加。Romeis等[20]发现,低渗胁迫能够诱导烟草钙依赖蛋白激酶基因NtCDPK2的磷酸化及其转录产物的增加。Yuasa等[21]在半咸水轮藻(Lamprothamniumsuccinctum)中发现了钙依赖蛋白激酶,并证明其参与了低渗条件下细胞体积的调节。而有关盐藻钙依赖蛋白激酶基因在该方面的研究尚未见报道。
本试验采用Real-time PCR技术分析了盐藻在高盐胁迫条件下的表达模式。研究发现,在正常生长条件下盐藻DsCDPK基因的表达量很低,当受到3 mol/L NaCl胁迫时,DsCDPK基因的表达量明显升高,胁迫1 h时表达量达到最高。说明盐藻DsCDPK基因受盐胁迫诱导。Harmon等[22]研究表明,钙依赖蛋白激酶可调节许多底物包括转录因子、代谢酶类、离子通道和离子泵、骨架蛋白等。推测钙依赖蛋白激酶可能调控盐藻在高盐胁迫下迅速的体积变化、离子的转运和细胞增殖等,在盐藻应答高盐胁迫的生理过程中可能发挥着非常重要的作用。
3.2 钙依赖蛋白激酶的原核表达与纯化
在前期工作中,克隆到了一种钙依赖蛋白激酶基因[23],为进一步研究该蛋白激酶的性质及功能,需要获得高纯度的可溶性蛋白。由于大肠杆菌表达系统具有易于培养和控制、操作简单、表达蛋白产量高、需时短、试验成本低、表达产物易于纯化等优点,在科研和生产领域已成为最常用的表达外源蛋白的宿主[24]。本研究中融合蛋白的表达采用原核表达系统,以大肠杆菌BL21为表达菌,将构建的原核表达载体pET-32a-DsCDPK导入受体菌,融合蛋白在大肠杆菌中获得了成功表达。但外源基因在宿主细胞的表达水平,不仅依赖于宿主本身,还受诱导条件、目的基因核苷酸序列组成、蛋白质的性质等因素的影响。有研究报道,如果目的基因的GC含量大于70%,可能会降低其在宿主细胞中的表达水平[25-27]。通过Bioedit软件分析发现,DsCDPK基因开放阅读框基因序列的GC含量为54.52%,对表达水平的影响不大。有研究表明,亲水性的蛋白质较疏水性的膜蛋白更易表达[28-29]。生物信息学分析发现,DsCDPK蛋白为亲水性蛋白质,这将提高其表达可溶性蛋白质的效率。经过反复摸索诱导条件,最后确定DsCDPK基因最适表达条件为:温度28 ℃,IPTG浓度0.6 mmol/L,诱导培养时间4 h。将获得的融合蛋白进行可溶性分析,发现在破碎细胞的上清液中表达量较多,而在包涵体中表达量较小,结果很理想。
为获得高纯度的融合蛋白,将收集的可溶性蛋白经His60镍柱纯化试剂盒纯化,能识别His标签的融合蛋白被吸附在柱子上,再经过洗脱,得到纯度较高的融合蛋白。为进一步确定得到的融合蛋白是否为目的蛋白,对纯化的融合蛋白进行Western杂交检测。结果显示,融合蛋白与羊抗鼠IgG单抗进行了特异性的结合,并且具有良好的免疫学活性,初步证明纯化的蛋白就是带有His标签的钙依赖蛋白激酶。后续的工作将利用获得的DsCDPK蛋白制备抗体,应用免疫共沉淀技术、液相二级质谱技术和生物信息学方法筛选出与钙依赖蛋白激酶相互作用的蛋白质,构建蛋白质相互作用网络,在蛋白质组学的水平上研究钙依赖蛋白激酶的功能,这将为进一步阐明钙依赖蛋白激酶在盐藻应答盐胁迫信号传递途径中的作用提供新的理论依据。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
基层中医药(2022年4期)2022-07-22
局解手术学杂志(2022年5期)2022-05-30
中国动物保健(2022年2期)2022-05-05
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
三农资讯半月报(2020年3期)2020-03-09
山西农业大学学报(自然科学版)(2020年1期)2020-03-04
科学大观园(2019年23期)2019-09-10
