
杆状体肌病新发突变临床特征及基因诊断
2019-03-28 06:44:40牛焕红成胜权
山西医科大学学报 2019年3期
牛焕红,成胜权
(空军军医大学第一附属医院儿科,西安 710032;*通讯作者,E-mail:quanyi@fmmu.edu.cn)
杆状体肌病(nemaline myopathy,NM)是一种罕见的先天性肌病,因在患者肌肉纤维中发现大量杆状体结构而得名,发病率报道不一,呈常染色体显性遗传(AD)/常染色体隐性遗传(AR)及散发。目前报道大多通过骨骼肌肌肉活检病理诊断,肌细胞胞浆中的杆状体是特征性肌肉病理改变[1]。本文报道的患儿经过基因检测确诊,该突变位点目前国内尚未见报道,且为新发突变,现将其临床特征及其基因诊断特点总结如下,提高临床医生对本病的认识。
1 资料与方法
1.1 病例介绍
患儿,男,2岁3个月,主因发育迟缓、肌无力2年余于2018年5月就诊我院。患儿于1月龄时即发现颈项软,不能抬头;1岁2个月时仍不会翻身、独坐、爬行、站立,一直以脑性瘫痪诊治;现回访2岁6个月可扶站、扶走,智力语言发育大致正常;患儿系第2胎第2产,足月剖腹产,出生体重2.7 kg,否认窒息缺氧史。家族史无特殊记载。查体:体重7.5 kg,脸型瘦长,表情淡漠,脊柱后凸(见图1),前囟闭合,头围47 cm,嘴唇稍厚,膨胀型嘴,高腭弓(见图2),四肢肌力3级,肌张力低,双侧上、中、下腹壁反射(++),双侧肱二头肌、肱三头肌腱反射(+),双侧膝、跟腱反射未引出,双侧巴宾斯基征未引出。无震颤、手足徐动、舞蹈样动作及感觉异常等。
辅助检查:血、尿、便常规、肝肾功、心肌酶谱、离子五项、血糖、血氨、血乳酸、血尿有机酸代谢筛查、染色体核型、心脏彩超、头颅核磁(MRI)均正常;双侧腓总神经、胫神经、正中神经、尺神经运动神经传导速度(MCV)大致正常;双眼视觉诱发电位(FVEP)、双耳听觉诱发电位(BAEP)未见异常;盖塞尔智能发育测验(1岁2个月时):动作能发育年龄(DA)2.8月、发育商(DQ)19.8分,应物能DA 8.7月、DQ 62分,言语能DA 9.3月、DQ 66分,应人能DA 10月、DQ 59分。
1.2 方法 基因测序
患儿生后不久即出现明显的肌力、肌张力减低,充分告知患儿家长并签署知情同意书后对患儿及其父母、姐姐各抽取3 ml全血,置EDTA抗凝管,送北京海思特医学检验实验室行基因测序,采用安捷伦外显子芯片捕获+高通量测序法,进行医学外显子5 000种疾病筛查。
2 结果
2.1 高通量测序结果
患儿1号染色体(chr1):229567822位点存在727(鸟嘌呤)G>(腺嘌呤)A的杂合突变,见表1。
2.2 sanger验证结果
患儿一人存在c.727G>A突变,父亲、母亲、姐姐无突变(见图3,4)。
2.3 对应数据库报道疾病
该样本在ACTA1基因外显子区域发现一处杂合突变(见表2),该突变HGMD数据库报道疾病为NM,家系验证结果显示该突变在父母双方均未检出,考虑为新发(De novo)突变。
表1杆状体肌病患儿高通量测序结果

突变基因转录本Exon编号核苷酸变化氨基酸变化染色体位置深度测序ACTA1NM-001100exon5c.727G>Ap.Glu243Lyschr1-22956782251/86(0.63)
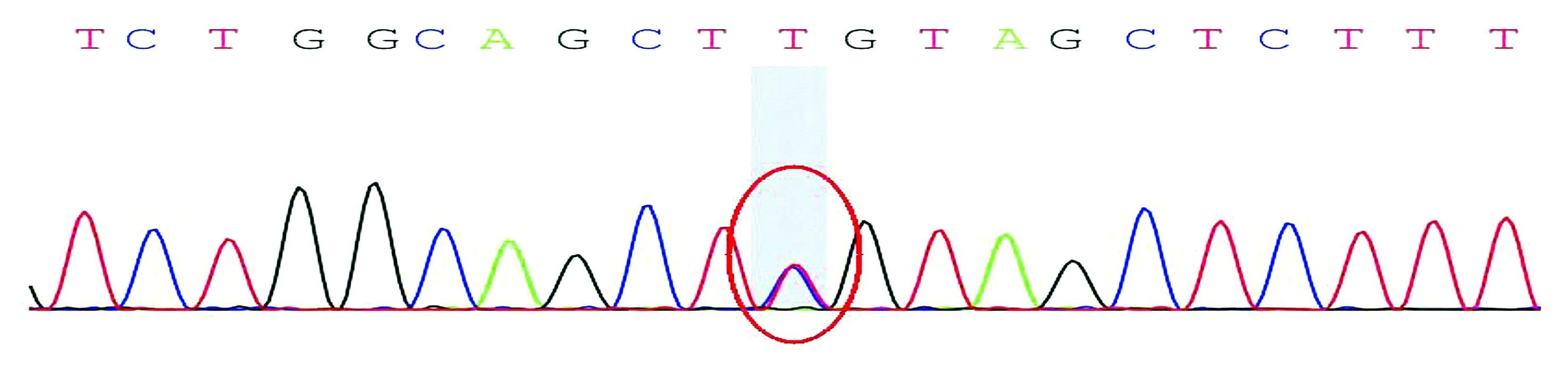
图3 杆状体肌病患儿sanger验证
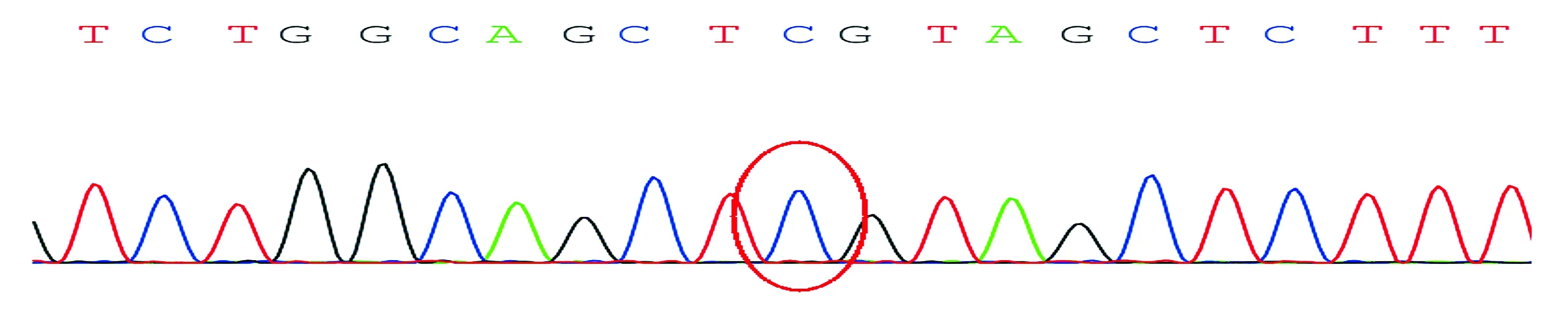
图4 患儿父亲sanger验证
表2杆状体肌病患儿HGMD专业版数据库以及ACMD分级

基因突变位点HGMDpro报道生物学危害性ACMD分级ACTA1c.727G>ADM:杆状体肌病可能有害(SIFT和Polyphen评估)-
3 讨论
3.1 NM的致病基因
NM属于罕见的先天性肌病,其主要病因为编码细肌丝相关蛋白的基因突变[2]。目前已发现可引起NM的致病基因包括ACTA1、TPM3、TPM2、KBTBD13、NEB、TNNT1、CFL2、KLHL40、KLHL41、LMOD3、MYPN和MYO18B等10余种,NEB基因变异是NM最常见的原因,约占已发现突变基因的50%[3]。而ACTA1为NM第2常见致病基因,约占已发现突变基因的15%-25%[4],也是目前唯一已知与核内杆状体相关的基因。ACTA1编码骨骼肌α-肌动蛋白,该蛋白可存在于1型和2型肌纤维中,目前已发现200余种ACTA1突变,主要为错义突变,多为AR,少数为AD。本文病例的ACTA1基因外显子区域发现1处杂合突变,染色体定位1q42,致c.727G>A(鸟嘌呤>腺嘌呤),从而导致编码α-肌动蛋白的氨基酸发生改变,p.Glu243>Lys(谷氨酸>赖氨酸),根据HGMDpro数据库报道为致病,报告疾病为杆状体肌病,家系验证结果显示该突变在父母双方均未检出,考虑为新发(De novo)突变,但不排除父亲或母亲存在生殖细胞突变嵌合体可能。根据SIFT和PolyPhen评估生物学危害性为可能有害,理论上此突变导致疾病发生的可能性大,可能为患者表型相关的致病突变。
3.2 NM的临床特征
根据2000年欧洲神经肌肉病委员会国际协作组对NM的临床分型,主要分为以下6型:①先天重症型(16.1%):出生时即有严重的肌无力、肌张力低下,严重的呼吸功能障碍,多早期死亡,年龄不超过1岁。②先天中间型(20.3%):婴儿期起病,出生时可有自主呼吸运动,但儿童早期可发展为无法自主呼吸运动,无法独立行走和站立,11岁前借助轮椅是主要特征之一。介于先天重症型和先天轻症型之间。③轻症型(经典型)(46.1%):婴儿期/儿童期起病,主要以面肌、躯干肌无力,伴肌张力低下,运动发育迟缓,随年龄增长肌力会有增加,病情相对稳定,多数患者的生活质量不受影响,智力、心肌收缩力多为正常。④儿童起病型(13.3%):与轻症型类似,区别在于该型起病年龄为儿童晚期或青少年期。⑤成人起病型(4.2%):起病30-60岁,是一组异质性疾病,临床表现和疾病进展都有很大的差异,多为散发,急性或亚急性起病,病程进展加重较快,易累及呼吸肌,多预后不良。⑥其他形式的NM:少见,表现为心肌病、眼肌麻痹、异常肌无力分布、核内杆状体。根据患者致病基因及染色体定位的不同,所编码的蛋白质及临床表型不同,根据其对应的临床表型有助于判断其预后。如TPM2、CFL2基因突变,导致其分别编码的β原肌球蛋白、人肌动蛋白素2发生改变,均可导致先天轻症型NM发病;而ACTA-1、NEB、TPM3突变根据其染色体位点的不同所致NM可出现先天重症型、轻症型、其他各型等多种表型[2]。本文中患儿ACTA1基因突变,致其所编码的α-肌动蛋白发生改变,导致临床表型为轻症型,随年龄增长肌力增加,现可扶站扶走,智力与同龄儿相仿,预后较好。
3.3 NM的诊断与鉴别诊断
3.3.1 诊断 该病的诊断主要依靠骨骼肌肌肉活检,无论哪种致病基因所致,其特征性表现均为肌细胞胞质中可见高密度的杆状体,MGT染色可以见到最为典型的红染杆状体,不同部位的肌肉组织及不同肌纤维中杆状体数目不尽相同,故取材不同可能会漏诊,需二次或多次取材。尤其对于综合医院的患儿,由于条件限制肌肉活检很难实施,因肌肉活检术为有创检查且术后患儿活动受限、伤口护理及恢复需要时间。随着分子生物学技术的发展与应用,肌肉活检不再是唯一诊断的金标准,将会有越来越多的NM通过基因检测被报道。
3.3.2 鉴别诊断 临床对于有肌无力的患儿需要鉴别的疾病较多,如进行性肌营养不良、皮肌炎、脊髓性肌萎缩、脑性瘫痪等等,应根据其临床病史及查体做出初步定位定性判断,对于像该患儿出现全身软性瘫痪者,定位为下运动神经元及其以下病变,如能排除神经传导与神经肌肉接头处病变如重症肌无力后,考虑定位诊断位于脊髓前角的运动神经元或肌肉时,二者临床很难鉴别,若肌电图能发现巨大电位、纤颤电位、失神经电位、高幅多相运动电位,可能为运动神经元病;若发现短暂、低幅多相运动电位可能为肌肉病变,然而,要做到这一点实属不易,需要高年资有丰富经验的儿童肌电图医生来实现,最终二者的鉴别需要骨骼肌病理活检或相应的基因检测结果方可确诊。
本病尚无有效治疗方法,有研究[5]报道,采用马法兰或者免疫抑制剂如环磷酰胺和甲基强的松治疗能改善部分病人的临床症状,但尚未经过大规模试验。随着分子生物学技术的发展与应用,NM的发病机制研究以及基因治疗将可能成为新的研究热点。
猜你喜欢
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:30
国际放射医学核医学杂志(2020年2期)2020-05-30 12:39:56
测绘工程(2019年6期)2019-09-21 07:46:00
中国中医急症(2019年10期)2019-05-21 07:20:42
科学之谜(2019年3期)2019-03-28 10:29:44
城市勘测(2018年6期)2019-01-03 09:07:54
橡塑技术与装备(2018年20期)2018-10-20 02:29:20
科学之谜(2018年8期)2018-09-29 11:06:46
现代检验医学杂志(2016年4期)2016-11-15 02:00:58
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
