
ABCC8基因突变致先天性高胰岛素血症性低血糖1例并文献复习
2019-03-27 07:25:20黄春玲韩彤妍
国际内分泌代谢杂志 2019年1期
黄春玲 韩彤妍
北京大学第三医院儿科 100191
新生儿低血糖症在临床上较常见,持续性或顽固性低血糖可导致低血糖脑病的发生。而先天性高胰岛素血症(CHI)是导致婴幼儿持续性、重度低血糖的最常见原因,也是导致新生儿低血糖脑病的重要因素[1-2]。其特征是与血糖水平不相符的胰岛β细胞不规律释放胰岛素[3]。1954年由MacQuarrie首次描述,当时诊断为婴儿特发性低血糖症,该病发病率在不同种族之间差异很大,在全世界的活产婴儿中发病率为1/15万~1/3万[4]。文献报道新生儿的发病率为1/5万~1/3万[5]。在近亲婚配的群体中,发生率高达1/2 500[6]。本文报道1例CHI性低血糖患儿的临床特点,并复习相关文献。
1 病例介绍
患儿,女性,生后32 h,于2014年11月22日因发现体温低、反应差2 h入院。患儿家属于入院前2 h发现患儿反应差,刺激后哭声弱,四肢活动少,嗜睡,吃奶差,伴呻吟,测体温34.5℃,无明显惊厥、呼吸困难、发绀等表现,为进一步诊治由门诊收入北京大学第三医院儿科。患儿系第3胎第2产,胎龄37+6周,母孕期合并妊娠糖尿病(糖耐量减低,饮食控制血糖在正常高限)、先兆子痫、霉菌性阴道炎,经阴道分娩出生,无宫内窘迫及生后窒息,无胎膜早破,羊水、胎盘、脐带无异常,出生体重3 390 g。生后母乳喂养,自行吸吮差,母乳量少,生后24 h内排尿2次,未排便。生后24 h开始喂配方奶,20~30 ml/次,3~4 h/次,吃奶尚可,排尿、排便增多,目前仍排胎便。出生后哭声尚可,哭闹时偶伴有口周青紫,但肌张力减低。
家族史:家族中无类似表现及疾病。姐1人,8岁,平时食量大,幼时需2 h吃一次奶。
查体:生命体征平稳,体重3 110 g(较出生体重下降8.3%),发育正常,面色红润,哭声弱,四肢动作少,四肢伸展。全身皮肤中度黄染,阳黄。前囟2 cm×2 cm,无膨隆及凹陷,张力不高。无先锋头及头颅血肿。双眼凝视,双侧瞳孔等大正圆,对光反射灵敏。呼吸平稳,有呻吟,无口周发绀,三凹征阴性,双肺呼吸音稍粗,无啰音,心音有力,律齐,胸骨左缘2~3肋间可闻及2/6级收缩期杂音,无传导。四肢末梢凉,毛细血管再充盈时间3 s。腹软,肠鸣音正常,肝肋下1 cm,剑突下2 cm可触及,脾肋下未及。四肢肌张力减低,腘角>90度,原始反射未引出。
实验室检查:血、尿、便常规正常,C反应蛋白19 mg/L,后复查正常。肝功能、肾功能、电解质正常。乳酸、血氨水平正常。脑脊液常规、生化正常,涂片未见异常。血培养正常,脑脊液培养正常。凝血功能大致正常,术前免疫8项正常,甲状腺功能正常。入院后血糖最低1.4 mmol/L,空腹胰岛素24.4 mU/L,同期空腹血糖3.9 mmol/L,胰岛素/血糖0.34>0.3,考虑CHI,C肽水平在正常范围,生长激素水平在正常范围。血、尿氨基酸、有机酸代谢筛查未见异常。尿酮体阴性,染色体及微缺失检查未见异常。
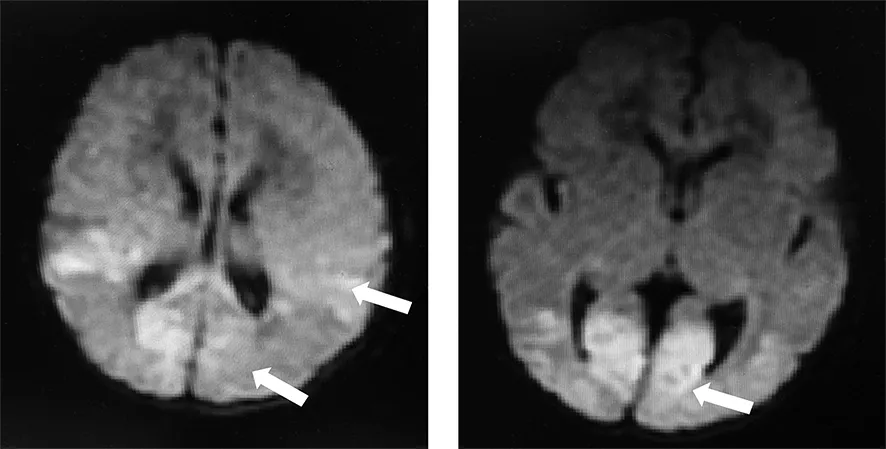
影像学检查:胸片未见异常、腹部超声未见异常。头颅超声:双侧弥漫性脑实质损害,双侧脑室内少量出血(基本稳定)(图1)。头颅核磁:双侧颞顶枕叶急性脑梗死可能,双侧横窦静脉血栓形成可能(双侧顶叶近侧脑室旁。双侧枕叶可见斑片状长T1长T2信号影,双侧横窦内可见异常短T1短T2信号改变;DWI:双侧颞顶枕叶可见较多片状高信号影)(图1)。头颅MRA:双侧大脑前动脉及大脑后动脉远端管壁欠光滑。心脏超声:心房水平左向右分流(多发房间隔缺损)三尖瓣少量反流。脑电图(d3):异常脑电图。背景活动波率偏快;变异性缺失;右侧额、中央顶枕颞区尖波发放,左侧少见。
为进一步明确病因,行相关基因检查发现患儿ABCC8基因错义杂合突变:c.4477G>A,导致氨基酸改变:p.R1493W,精氨酸(Arg,R)>色氨酸(Trp,W)。家系调查未发现患儿家族中有类似疾病的成员,进一步完善其家族此基因检查,患儿父亲及姐姐也发现类似基因突变(图2),无临床表现,血糖正常,其母亲无类似突变。

2014年11月22日入院后约0.5 h,患儿出现惊厥,表现为双眼凝视,四肢强直抖动,约1 min缓解,查血糖低(1.4 mmol/L),给予静脉推注10%葡萄糖后复测血糖上升,逐渐上调糖速,静脉输注葡萄糖维持速度5.5 mg/(kg·min),患儿反应好转、肌张力正常,同时给予开奶,逐渐加奶至100 ml/次,患儿血糖可维持正常,生后14 d出院。
2 讨论
高胰岛素血症性低血糖症的临床表现不典型,不易被识别,症状不典型,为诊断带来困难。本病区别于其他新生儿常见低血糖症的临床特点为:严重的难以纠正的低血糖;进餐后也难以维持2 h以上的正常血糖水平;在给予葡萄糖静脉点滴时,所需葡萄糖速度较高;低血糖时血酮体水平在正常范围内,尿酮体阴性;不伴有酸碱平衡紊乱等。高胰岛素血症性低血糖诊断需在低血糖状态下立刻进行血液学检查,但溶血会致红细胞内胰岛素降解酶释放,故检测胰岛素水平并不总是可行。这时需根据胰岛素分泌过量所致的其他表现来诊断,例如游离脂肪酸、3-羟基丁酸水平降低;另外,可根据给予胰高血糖素后出现不相符的血糖反应来判断。此外,临床上无法根据胰岛素明确病因时,可以进一步进行基因或其他代谢物质检查[7]。
基因突变是CHI的主要原因,基因检查不仅可以进一步确诊此病,而且为进一步治疗提供依据。参与胰岛素分泌的重要蛋白编码基因突变是高胰岛素血症的最常见原因[8-9]。现已知 9 种致病基因可引起 CHI,包括与离子通道病相关的ATP结合盒转运蛋白C8(ABCC8)、钾离子内向整流通道蛋白J亚单位11号成员(KCNJ11)基因及与代谢性疾病相关的谷氨酸脱氢酶1(GLUD1)、葡萄糖激酶(GCK)、羟酰辅酶A脱氢酶(HADH)、溶质载体家族16单羧酸转运蛋白成员1(SLC16A1)、解耦联蛋白2(UCP2)、肝细胞核转录因子(HNF)4α和HNF1α基因[10-11]。其中ABCC8基因突变是CHI最常见的致病基因,ABCC8基因位于染色体11p15.1,由39个外显子组成,长度超过100 kb,编码1 582个氨基酸,核酸大小约177 000。
本例患儿主要表现为反应差、体温低、惊厥,头颅核磁提示颞顶枕叶损伤,诊断低血糖脑病明确;本例为复发性严重性低血糖,住院期间虽维持葡萄糖速度不高,但喂奶需频繁、量大才能维持血糖稳定。空腹胰岛素24.4 mU/L,同期空腹血糖3.9 mmol/L,胰岛素/血糖0.34>0.3,考虑CHI,C肽和生长激素水平在正常范围,血、尿氨基酸和有机酸代谢筛查未见异常;染色体及染色体微缺失检查未见异常。为明确病因,行相关基因检查发现,患儿ABCC8基因杂合突变c.4477G>A,使蛋白的第1 493位氨基酸由精氨酸(Arg,R)变为色氨酸(Trp,W)。患儿父亲及姐姐也有类似基因突变,其母亲无类似突变,考虑为父系常染色体显性遗传可能性大。国外研究资料显示,父系遗传的ABCC8基因突变包括两种可能,即父系遗传的常染色体显性遗传基因突变和常染色体隐性遗传基因突变的特殊类型。前者父亲和患儿均携带同样的突变,患儿母亲突变位点的基因型正常,此类患儿临床症状轻重不一,起病年龄偏早[10]。后者多存在钾通道构成的异常,常导致局灶型ATP敏感性钾通道型先天性高胰岛素血症(KATP-CHI),病情较重,多于新生儿期发病,需要大量的葡萄糖维持血糖水平的正常,多对二氮嗪等药物治疗无效,需行不同程度的胰腺切除术来控制低血糖的发生[11]。
本例患儿未经药物治疗血糖控制满意,未再出现惊厥等神经系统异常,随访至近2岁,智力及运动发育与同年龄儿童无明显区别。葡萄糖是新生儿期脑组织代谢的唯一能源,新生儿脑组织葡萄糖的需要量大,但储存极少。CHI治疗最主要的目的是维持血糖水平正常,避免造成神经系统损伤。葡萄糖输注速率需提高至 15~20 mg/(kg·min)以上方可维持血糖在正常范围,或低血糖反应频繁出现,则需使用药物和手术治疗[12]。大部分患者初期都需要高速率葡萄糖持续输注以维持血糖稳定,当高速率葡萄糖持续输注仍不能维持血糖水平正常时,可考虑加用胰高血糖素或生长抑素治疗。治疗首选药物为二氮嗪,但其起效慢、反应不确定,难以控制低血糖,因基因类型及遗传方式影响治疗效果,所以应尽早完善基因检查,指导CHI的临床治疗用药。但目前仍有约50%的CHI尚未发现具体的基因突变[13],且其基因突变的遗传方式并不严格遵守经典遗传模式[14],即使检测到基因突变,也不一定能准确代表CHI的基因突变类型或明确其遗传方式,同种基因突变可能有不同的临床表型,病变类型也可能不同[15]。因而在血糖控制和完善基因检查后仍应进行二氮嗪试验性治疗,将基因结果结合治疗反应、临床表现、实验室检查、影像学检查综合判断,才能为CHI制定最佳的远期临床管理策略[16]。
本病的预后取决于患者所患的CHI类型及严重程度,最严重的并发症是低血糖脑病。甚至可遗留新生儿永久性脑损伤:如认知障碍、视觉障碍、枕叶癫痫、脑瘫等后遗症等。苏畅和巩纯秀[17]对15例CHI患儿进行分析,随访3例(二氮嗪治疗2例、胰腺次全切除1例)智力正常,2例放弃治疗后死亡,余10例均有中、重度智力低下,低血糖发作无缓解。本文患儿现3岁,未遗留明显的后遗症。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国甜菜糖业(2020年1期)2020-12-07 07:54:30
饮食保健(2019年2期)2019-01-17 05:35:44
江苏卫生保健(2018年7期)2018-07-31 08:35:34
小学生导刊(2018年13期)2018-06-29 03:49:00
妇女之友(2016年11期)2017-01-20 20:02:31
哈尔滨医药(2016年3期)2016-12-01 03:58:29
中国乳业(2016年4期)2016-11-07 09:50:27
肿瘤影像学(2015年3期)2015-12-09 02:38:50
