
CO2在三聚氰胺酚醛纤维上的吸附分离
2019-03-06 00:45:44,,,,,
燃料化学学报 2019年2期
, , , , ,
(浙江师范大学 “先进催化材料”教育部重点实验室, 浙江 金华 321004)
研究高效CO2吸附材料对于CO2捕集与分离、实现碳资源循环利用具有重要意义[1],迄今已有较多CO2吸附剂被报道,例如:Li基高温CO2吸收剂[2]、活性炭[3-5]、分子筛[6]、介孔SiO2[7]、胺基功能化介孔材料(AFMs)[8-11]、金属有机骨架材料(MOFs)[12-14]及多孔有机聚合物(POPs)[15-18]等。胺基多孔材料是近年来报道较多的一类CO2吸附材料,借助于胺基对CO2的高亲和力,胺基多孔材料显示了良好的CO2吸附性能[19,20]。Schwab等[21]报道了以三聚氰胺(密胺)和苯二甲醛同系物为原料,通过Schiff碱反应制得密胺基微孔聚合物材料(MB-MOPs)。得益于其丰富的微孔结构和高含量的胺基,MB-MOPs表现出良好的CO2吸附性能[22,23]。但三聚氰胺和对苯二甲醛的Schiff碱缩合反应需要在惰性气氛中通过溶剂回流进行,这限制了MB-MOPs的合成及应用。
最近,Long课题组利用三聚氰胺、间苯二酚和甲醛为前驱体合成出密胺基微球,利用间苯二酚、甲醛和三聚氰胺共交联形成的网络结构分散胺基基团,合成的密胺基微球比表面积接近为零,在298 K、100 kPa下CO2吸附量为1.8 mmol/g[24,25]。随后作者课题组[26]采用简单的水热缩聚反应合成出三聚氰胺-间苯三酚-甲醛纳米纤维(PMF),其比表面积为42 m2/g, 298 K、118 kPa下的CO2吸附量为1.12 mmol/g。考察了N2气氛中炭化PMF,发现800 ℃下炭化后的PMF纳米纤维有较好的CO2吸附性能,298 K、100 kPa下的CO2吸附量提高到2.1 mmol/g。
本研究考察了温度对PMF合成的影响,并采用合适的吸附模型分别对CO2和N2在不同温度下的吸附平衡等温线进行联合拟合,计算其在PMF上的等量吸附热;采用穿透柱技术研究CO2-N2双组份气体在PMF上的吸附分离性能;并研究多种气氛中热处理对PMF结构和CO2吸附分离性能的影响。
1 实验部分
1.1 三聚氰胺酚醛纤维的制备
将2.21 g间苯三酚溶于20 mL去离子水中,搅拌溶解,滴加4.25 g甲醛溶液(37%),升温至313 K搅拌100 min,得到的混合液记为体系1。另将2.21 g三聚氰胺加入到15 mL去离子水中,搅拌均匀,滴加4.25 g甲醛溶液(37%),升温至343 K搅拌至溶液澄清,得到体系2。将体系2加入体系1中并转至反应釜内衬中,搅拌30 min后放入反应釜中,分别在298、353、393和423 K下反应24 h,样品记为T1、T2、T3、T4。用去离子水对产物进行抽滤、洗涤至滤液澄清,333 K下干燥24 h得到最终产物。
1.2 三聚氰胺酚醛纤维的后处理
选用T3样品进行后处理,将T3填充到内径为10 mm的不锈钢反应器中,分别在N2、H2、N2+5%水蒸气和N2+15%水蒸气下,从室温以8 K/min升温至873 K,保持5 h,即得到处理后样品,分别命名为T3-N2、T3-H2、T3-5%H2O和T3-15%H2O。
1.3 样品表征及吸附性能评价
样品形貌是用Hitachi S-4800型扫描电子显微镜(SEM)拍摄的,加速电压为5 kV,样品真空喷金后测试。透射电子显微镜(TEM)照片在2100 JEOL型透射电镜上获得,工作电压为200 kV,将样品经乙醇稀释并超声处理后滴加到覆有碳薄膜的Cu网上制样。在Nicole NEXUS 670型红外光谱仪上收集红外谱图,室温下采用KBr压片制样,记录4000-500 cm-1。77 K下,N2吸脱附等温线在美国Micromeritics公司ASAP 2020物理吸附仪上获得,分析前样品在393 K下真空脱气12 h,比表面积由BET(Brunauer-Emmett-Teller)法计算得到,微孔孔容是用t-plot模型测得,以相对压力p/p0=0.97时的吸附量计算得到总孔容。单组分气体吸脱附等温线在Micromeritics ASAP 2020物理吸附仪上获得,真空393 K下将样品脱气12 h,在压力0.01-118 kPa下进行吸附测试,温度为298-323 K。穿透柱实验在自制的装置上进行,在线质谱仪检测流出气体组成。实验前用He流在393 K下纯化样品12 h,进行实验时通入He-CO2-N2(4∶2∶2, 体积比)混合气,气体总流量为8 mL/min。在相同条件下,用相同体积SiO2进行空白实验。利用赛默飞世尔科技公司ESCALAB 250Xi型X射线光电子能谱(XPS)测定样品中C、N、O的状态及含量,对N 1s测试结果拟合分峰,确定样品中氮种类。
2 结果与讨论
2.1 不同温度下合成PMF
分别考察了298、353、393和423 K下间苯三酚、三聚氰胺、甲醛的缩聚反应,结果发现,不同合成温度下均可得到粉末状PMF。图1为不同温度下合成的PMF的SEM照片,由图1可知,PMF样品均具有纤维形貌,T1样品纤维细长并交错在一起,可能是由于室温下缩聚反应不完全,样品在抽滤烘干过程中继续反应使得纤维黏结到一起。随着温度的升高,T2、T3样品纤维逐渐分散,并且规整度有所提高。进一步升高反应温度到423 K,T4样品纤维长度变短,直径变粗。

图 1 PMF样品的SEM照片
图2显示不同温度下合成的PMF纤维呈束状,并交错在一起,随着温度升高,纤维束逐渐分离,纤维直径从25 nm逐渐增大到40 nm,但经进一步升高温度所合成的T4样品,纤维规整度有所降低,直径粗细不匀。

图 2 PMF样品的TEM照片
图3为PMF样品的红外光谱谱图。

图 3 PMF样品的红外光谱谱图
由图3可知,1555和1490 cm-1处为三聚氰胺中C=N的吸收峰,1340 cm-1处为其C-N吸收峰,说明合成的PMF材料中含有三聚氰胺的三嗪环;3400、810 cm-1处分别为N-H的伸缩振动峰和弯曲振动峰,说明原料中的三聚氰胺参与了缩聚反应进入到PMF材料中;2970 cm-1左右对应苯环中氢的伸缩振动峰,说明材料中有间苯三酚参与反应;1680 cm-1处未见醛基的C=O伸缩振动峰,表明PMF材料中没有游离的醛基。较低温度下制备的T1样品在1555、1490 cm-1处的吸收峰明显较弱,可能是因为三聚氰胺在反应中参与程度低。77 K下N2吸脱附等温线见图4,样品均显示出典型的Ⅱ型等温线特征,在相对压力为0.95-0.99,观察到吸附量迅速上升,并出现滞后环,这是由于纳米纤维聚集在一起形成二次孔。表1为PMF样品的孔结构数据。T1样品具有最大的孔容,这可能是由于T1样品纤维交错连接到一起形成更多的二次孔。393 K下合成的T3样品比表面积最大(64 m2/g),更为重要的是,T3样品的微孔比表面积和微孔孔容均最大,分别为16 m2/g和0.008 cm3/g。丰富的微孔结构对于气体吸附具有重要作用。

图 4 77 K下PMF的N2吸脱附曲线

表 1 PMF样品的孔结构数据
采用体积法测试了CO2在PMF上的吸脱附平衡等温线,具体见图5。由图5可知,吸脱附等温线在100-118 kPa基本重合,在100 kPa以下,存在不同程度的脱附滞后现象,但最终可以闭合,说明降低压力能够实现CO2从PMF上脱附。在298 K下,PMF均有较高的CO2吸附量,吸附量随压力增加而增加。结合表1的孔结构分析,可以看出CO2吸附量与材料的微孔容和比表面积密切相关。T3表现出较好的CO2吸附性能,吸附量为1.83 mmol/g(118 kPa、298 K)。

图 5 298 K下CO2在PMF上的吸脱附等温线
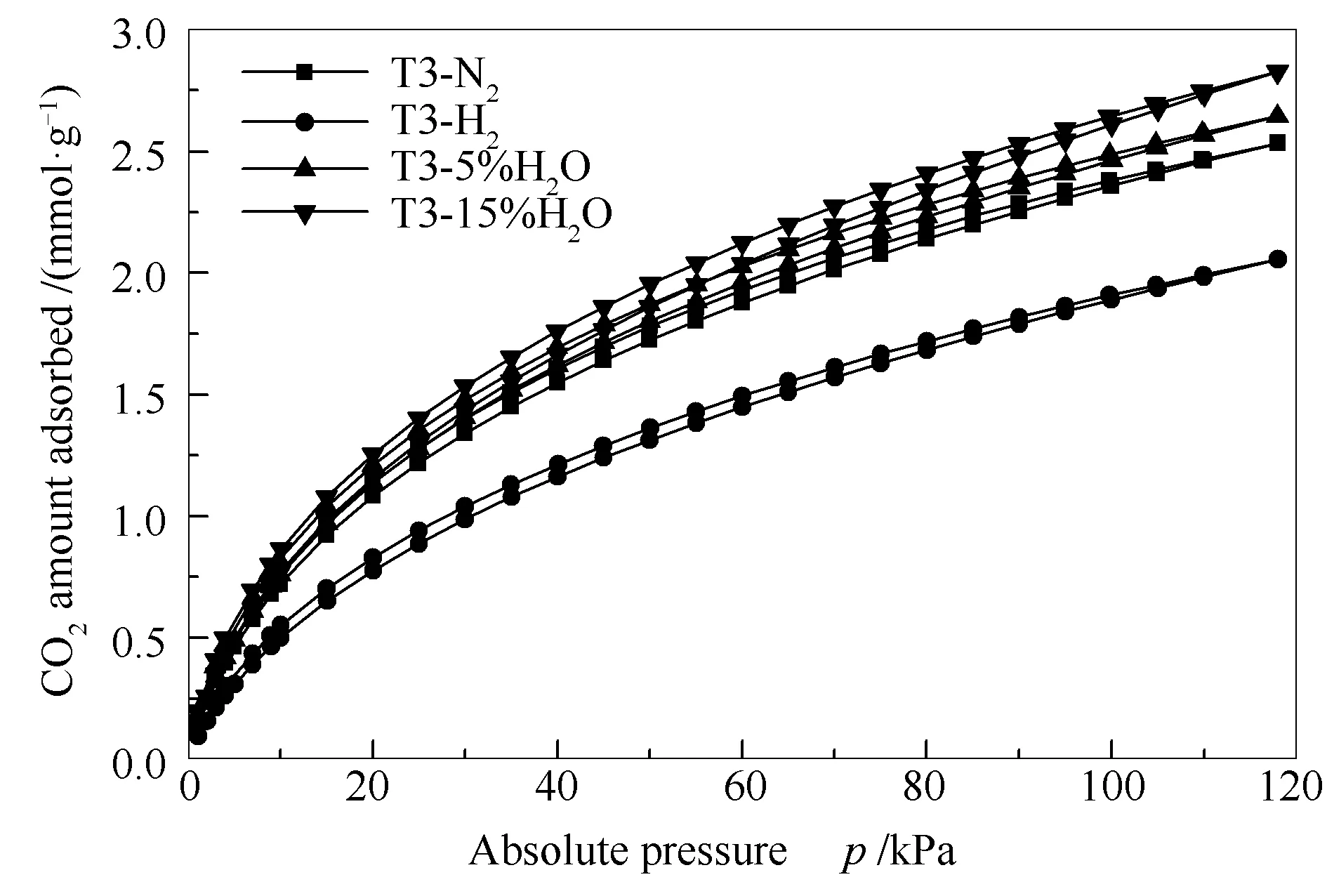
分别测试了不同温度下(298、310、323 K)T3样品的CO2、N2吸附等温线。一般认为CO2在胺基多孔材料上存在两种吸附位点:碱性胺基位1和微孔吸附位2,而N2只有一种吸附位点:微孔吸附位,因此,分别采用双位Langmuir(DSL)模型和单位Langmuir(SSL)模型拟合CO2、N2吸附等温线[23]。采用DSL模型联合拟合CO2吸附等温线。拟合曲线较好地符合实验点,吸附平衡常数均随着温度的升高而降低,第一吸附位的平衡常数b1大于第二吸附位平衡常数b2,说明碱性胺基位1对CO2的吸附强于微孔吸附位2;而q1,sat 通过穿透柱实验测试不同压力下双组分气体CO2-N2在T3上的分离性能,结果见图6。 图 6 298 K,不同压力下CO2-N2在T3上的穿透曲线和解吸曲线 观察图6中所有穿透曲线,弱吸附气体N2均先从柱子中穿出,并有roll-up现象,CO2随后流出,从而实现对双组分气体的分离,根据穿透曲线形状,可以判定为基于热力学选择性的分离[27]。随着压力的升高,气体在柱子中的停留时间延长,穿透窗口变宽,从穿透曲线计算的CO2吸附量也相应增加,但选择性逐渐降低。待CO2和N2混合气体从柱子穿透后,切换气体为He进行吹扫,发现N2比CO2先从柱中脱附出来,在298 K下,经长时间吹扫也可将CO2脱附出来。这说明实际工业操作中,可通过气体吹扫或降低压力实现吸附剂再生。 为进一步提高PMF的CO2吸附性能,对PMF样品分别在不同气氛中进行高温热处理。图7为不同气氛中热处理后PMF样品的SEM照片。由图7可知,N2和H2气氛中处理后的样品依然保持纤维形貌,而在5%和15%水蒸气中处理后的纤维之间开始黏连到一起,形成连续的网状结构。这可能是由于在高温水蒸气存在下,PMF纤维表面基团进一步反应使得纤维黏结到一起。 图 7 不同气氛中热处理后PMF样品的SEM照片 图8为77 K下热处理后样品的N2吸脱附曲线。 图 8 77 K下热处理后样品的N2吸脱附曲线 由图8可知,热处理后样品的吸附量显著提高,其吸脱附等温线为典型I型等温线,在低压区有较高的氮气吸附量,表明PMF在不同气氛中热处理均可提高其微孔含量。处理后样品的孔结构数据见表2,微孔孔容和比表面积显著增大。其中,H2气氛中热处理后样品的比表面积和微孔孔容最大,分别为490 m2/g和0.16 cm3/g。间苯三酚、三聚氰胺和甲醛经缩合反应形成亚甲基胺桥链、醚桥链及亚甲基桥链,在炭化过程中,有机骨架中的醚桥链和亚甲基桥链不断地断裂,芳香环重新结合,逐渐形成类石墨片层,产生丰富的微孔结构,亚甲基胺桥链在高温下分解会产生NH2、NH和H等自由基,这些自由基与炭表面反应生成各种含氮官能团,如:-NH2、-CN、吡啶型N、吡咯型N等[28,29]。氢气气氛中可以抑制炭化后产物过度石墨化,因而具有更大的比表面积。在水蒸气存在下,有机物会热解释放出低分子的烃类、CO2及H2等气体,同时通过芳构化形成芳环,进而缩聚成平面的盘状稠环芳烃大分子,因而会具有更高比例的吡咯型N。 表 2 热处理后PMF的孔结构数据 采用体积法测试处理后样品的CO2吸脱附等温线,具体见图9。 图 9 298 K下CO2在热处理后样品上的吸脱附等温线 由图9可知,与处理前样品相比,CO2吸附量均有提高。H2气氛中处理后得到的T3-H2样品具有最大的比表面积和微孔孔容,但其CO2吸附量并非最高,相比处理前,其CO2吸附量并未有大幅提高。而经15%水蒸气处理后的T3-15%H2O样品CO2吸附量最大,达到2.83 mmol/g(298 K、118 kPa)。总压200 kPa、298 K下的穿透曲线也表明,CO2在T3-15%H2O样品柱内的较T3样品具有更宽的穿透窗口,说明15%水蒸气热处理进一步提高了其CO2吸附分离性能。 对样品进行XPS表征可知,T3-H2样品的C 1s、N 1s和O 1s含量分别为89.9%、5.7%和4.4%,而T3-15%H2O样品的C 1s、N 1s和O 1s含量分别为84.3%、8.1%和7.6%,高温热处理后PMF纳米纤维中N含量从24%显著降低到5%-8%[26],相较于孔隙率,PMF中的胺基对于CO2的吸附具有更大的贡献[24],这可能是高温热处理后样品的比表面积大幅增加,但其CO2吸附量仅从1.83 mmol/g增大到2.83 mmol/g(298 K、118 kPa)的原因。相比于T3-H2样品,T3-15%H2O样品具有较高的N/C。进一步对N 1s峰进行解析,根据N元素可能存在的五种状态[29]:氮化物(Nitride, 396.8 eV)、吡啶型N(Pyridinic, 397.6 eV)、胺基(Amine, 398.1 eV)、吡咯型N(Pyrolic, 399.5 eV)和石墨型N-O(Graphic N-O, 401.1 eV)对N 1s峰进行拟合,拟合结果见表3。T3-H2样品和T3-15%H2O样品中N的主要存在形式均为吡啶型N和吡咯型N,一般认为吡咯型N具有更好的CO2捕集性能[29],T3-15%H2O样品中吡咯型N含量为51.8%,略高于T3-H2样品。因此,在T3-15%H2O样品具有更高的N/C和吡咯型N含量的情况下,显示了更高的CO2吸附量。 表 3 XPS谱图N 1s分峰拟合结果 在不同温度下,通过三聚氰胺、间苯三酚和甲醛的水热缩聚反应制备了三聚氰胺酚醛纤维(PMF),393 K下合成的PMF具有较高的比表面积(64 m2/g)和较高的CO2吸附量(1.83 mmol/g,298 K、118 kPa),且对CO2有较高的吸附选择性,通过穿透柱实验发现CO2-N2混合气在PMF上可实现分离。873 K下PMF样品在N2、H2及水蒸气等多种气氛中处理后,比表面积和微孔孔容均有显著增加,在H2气氛中处理后的PMF样品的比表面积和微孔孔容分别可达490 m2/g和0.16 cm3/g。而在15%H2O中处理后,PMF纤维黏结到一起,其CO2吸附量可提高至2.83 mmol/g(298 K、118 kPa)。XPS表征结果显示经15%H2O处理后样品具有较高的N/C,且其中N的类型主要为吡咯型N。水蒸气气氛中热处理是提高三聚氰胺酚醛纤维CO2吸附量的有效途径。
2.2 PMF高温热处理




3 结 论
猜你喜欢
化工设计通讯(2024年1期)2024-04-08 02:50:52
能源化工(2022年3期)2023-01-15 02:26:43
中国特种设备安全(2021年4期)2021-10-13 06:42:14
净水技术(2020年12期)2020-02-16 11:26:12
北京航空航天大学学报(2017年9期)2017-12-18 07:12:18
宿州学院学报(2016年7期)2016-08-08 09:51:53
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:57
化工进展(2015年3期)2015-11-11 09:08:56
中学政史地·教学指导版(2014年10期)2015-02-02 08:59:38
无机化学学报(2014年5期)2014-02-28 17:31:40
