
rDNA 介导的乳酸克鲁维酵母整合型表达载体的构建及初步验证
2019-02-06 07:21孙海烨李由然顾正华丁重阳石贵阳
食品与生物技术学报 2019年10期
孙海烨,张 梁*,2,李由然,顾正华,丁重阳,2,石贵阳,2
(1.粮食发酵工艺与技术国家工程实验室,江南大学,江苏 无锡 214122;2.江南大学 工业生物技术教育部重点实验室,江苏 无锡 214122)
乳酸克鲁维酵母(Kluyveromyces lactis)能以乳酸为唯一碳源和能源,具有营养要求简单、生长速率快、分泌蛋白能力强、不会对蛋白高度糖基化、不产生内毒素等优点[1],作为美国FDA(Food and Drug Administration)认定的食品安全级酵母菌,到目前为止,已有上百种蛋白在该酵母中成功表达并实现规模化生产,涉及食品、生物和医药等领域[2]。K.lactis外源基因表达的载体主要包括游离型和整合型两类,前者如pKD1、pKRAS,拷贝数高但在无选择压力条件下易丢失,后者主要为当前应用最广泛的载体pKLAC1,稳定性高,但拷贝数相对较低且只能满足单个基因的整合表达[3-4]。此外,K.lactis 在外源蛋白分泌表达中仍存在瓶颈,其蛋白产量还有很大的提升空间。随着生物技术的广泛应用,酵母表达系统得到相应的发展,Dujon 等于2004 年完成K.lactis 的全基因组测序[5],有利于深入掌握该菌株的遗传信息并进行合理改造。研究者主要通过增加外源基因的剂量或共表达分泌途径中的辅助因子、分子伴侣等方式来提高外源蛋白的产量,而目前表达载体的选择较为局限,因此亟待构建合适的分子工具满足工业化的不同需求。
核糖体DNA(rDNA)是细胞核中编码核糖体RNA(rRNA)的脱氧核苷酸序列,由转录区和非转录区组成,具有高度保守性,以重复单元存在于真核生物基因组中[6]。以rDNA 为同源重组位点构建表达载体,在无选择压力条件下可以实现单个目的基因多拷贝或多个目的基因单拷贝稳定整合于基因组中[7-8],通过增加外源基因拷贝数或共表达辅助因子从而实现外源蛋白高效分泌表达的目的[9],且对宿主菌无不良影响[10],该策略已成功应用于酿酒酵母(Sacchormyces cerevisiae)[11-12]、毕赤酵母(Pichia pastoris)[9]、K.lactis[13-15]、产朊假丝酵母(Candida utilis)[16]、多形汉逊酵母(Hansenula polymorpha)[17-18]、解脂耶氏酵母(Yarrowia lipolytica)[19]、法夫酵母(Phaffia rhodozyma)[20]、Arxula adeninivorans[21-22]和产甘油假丝酵母(Candida glycerinogenes)[23]等酵母菌的异源表达。研究发现,整合位点在rDNA 单元中的位置、性质,表达盒的大小以及筛选标记的选择对整合效率和稳定性产生影响[6,11,14,18-19],从单拷贝到多达数十个拷贝均有报道,表明整合位点的选择导致宿主菌种基因拷贝数的差异。K.lactis 基因组中rDNA 有68 个重复单元,每个单元长度为8.6 kb 左右,由5S、26S、18S、基因内转录间隔序列(ITS)和外转录间隔区(ETS)组成[14,24]。目前,在K.lactis 中利用rDNA 介导同源重组尚处于初步阶段,研究发现因选取rDNA 单元中不同位置的同源整合序列,重组菌的整合拷贝数有很大差异,少则几个,多则达到120 个[13-15]。现阶段,对于rDNA 重复单元的结构、重要元件的作用以及分子重组机制等缺乏系统认识,需要进一步深入研究。
腺苷酸脱氨酶(AMP deaminase,EC 3.5.4.6)是一种氨基水解酶,催化腺苷酸脱去氨基产生肌苷酸和NH3,在嘌呤代谢循环中起着至关重要的作用,维持机体免疫力和腺苷酸能荷,作为工业生产中重要的酶已广泛用于食品、医药和农业等领域[25]。基于该酶稳定性高、易于定量检测的优点,以AMPD 为报告基因,并以实验室前期研究中的强启动子pScGAL7 为外源基因表达的调控元件,构建18S rDNA 介导的整合型表达载体pTRGA-amdS,利用RT-QPCR 测定重组菌外源基因的整合拷贝数[26],初步探究拷贝数与AMP 脱氨酶酶活的关系,并测定重组载体的稳定性,为进一步研究rDNA 单元在K.lactis 基因工程中的应用作了初步尝试,对具有广阔应用前景的K.lactis 进一步分子改造奠定基础。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株 文中所用质粒和菌株见表1。

表1 本研究中所使用的质粒和菌株Table 1 Plasmids and strains used in this study
1.1.2 引物 根据基因序列设计PCR 特异性引物(表2),委托金唯智公司合成。
1.1.3 培养基 LB 培养基(g/L):蛋白胨10.0,酵母粉5.0,NaCl 10.0,固体培养基添加20 g/L 琼脂粉,添加终浓度为100 μg/mL 的氨苄青霉素用于E.coli转化子的筛选;YEPD 培养基(g/L):蛋白胨20.0,酵母粉10.0,葡萄糖20.0,用于K.lactis、S.cerevisiae的培养;YEPDG 培养基(g/L):蛋白胨20.0,酵母粉10.0,葡萄糖10.0,半乳糖10.0;YEPG 培养基(g/L):蛋白胨20.0,酵母粉10.0,半乳糖20.0;YCB 固体培养基(g/L)[26]:酵母基础碳源11.7,1 mol/L pH 7.0 的KH2PO4-K2HPO4缓冲液30 mL/L(终浓度30 mmol/L),琼脂粉20,灭菌后加入0.5 mol/L 的乙酰胺10 mL/L(终浓度5 mmol/L),用于K.lactis 重组菌的筛选。
1.1.4 工具酶、主要试剂及仪器 限制性内切酶Kpn I、Xho I、Sal I、Hind III、Bgl II,美国Fermentas公司;SYBR Premix Ex TaqTMII、CIAP、T4 DNA 连接酶,大连Takara 公司;质粒小量提取、DNA 片段的纯化、回收试剂盒,pfu、taq 等DNA 聚合酶,杭州宝赛生物技术有限公司;氨苄青霉素(Amp),上海生工生物工程股份有限公司;酵母基础碳源(yeast carbon base,YCB),New England Biolabs 公 司;5’-腺苷酸,美国Sigma 公司。Multiporator 电转仪,德国Eppendorf 公司;Bio-Rad S1000 PCR 仪、Chemi Doc凝胶成像仪、CFX96 荧光定量PCR 仪,美国Bio-Rad公司;紫外分光光度计,美国UNICO 公司。
1.1.5 溶液配制 原始底物:取1 mL 0.08 mol/L NaHCO3溶液溶解13.9 mg 5’-腺苷酸。反应底物:原始底物用pH 6.0 的0.1 mol/L 琥珀酸钠-NaOH 溶液稀释400 倍至终浓度为0.1×10-3mol/L。
1.2 方法
1.2.1 基因克隆及质粒的构建 以质粒pT-AMPD为模板,扩增鼠灰链霉菌(Streptomyces murinus)来源并经密码子优化的AMPD 基因,Xho I 和Sal I 双酶切该片段,连接至经同样双酶切的载体pKLAC1,构建质粒pKLAC1-AMPD。以该质粒为模板,分别利 用pLAC4-AMPD-F/R、amdS-F/R 引 物,扩 增pLAC4-αMF-AMPD-tLAC4 片段和pADH2-amdSTT 片段,与质粒pMD 19T-simple 连接,构建质粒pTLA 和pT-amdS。
用酸性玻璃珠法提取酵母染色体基因组,以S.cerevisiae W303-1A 基因组为模板,pScGAL7-F/R为引物,扩增得到长度为301 bp 的pScGAL7 片段,Kpn I 和Hind III 双酶切连接至同样酶切的质粒pTLA,替换pLAC4,构建质粒pTGA。质粒pT-amdS经Bgl II 酶切后胶回收目的片段(2 527 bp)后连接至Bgl II 单酶切并经CIAP 去磷酸化的线性化质粒pTGA,经Dir1-F/R 引物验证正确的重组质粒命名为pTGA-amdS。
以K.lactis GG799 基因组为模板,分别以18S rDNA-3’-F/R 和18S rDNA-5’-F/R 为引物,扩增得到长度为1 199 bp 的18S rDNA-3’ 片段和562 bp的18S rDNA-5’片段,采用重叠延伸PCR 扩增得到18S rDNA-3’-18S rDNA-5’融合片段,与质粒pMD 19T-simple 连接,构建质粒pTR。
质粒pTGA-amdS 经Kpn I 酶切后胶回收目的片段(5 282 bp)后连接至Kpn I 单酶切并经CIAP去磷酸化的线性化质粒pTR,经Dir2-F/R 引物验证正确的重组表达载体命名为pTRGA-amdS。
1.2.2 重组载体电转化K.lactis GG799 Sac II 线性化重组表达载体pTRGA-amdS 后电转化K.lactis GG799,涂布YCB 平板,于30 ℃培养3~4 d,挑取单菌落提取染色体基因组,利用Int-F/R 引物进行PCR 验证,正确整合的重组菌可以扩增得到大小为2 032 bp 的片段。
1.2.3 内参基因GAPDH 的克隆及标准质粒的构建以K.lactis/pTRGA-amdS 染色体为模板,根据NCBI数据库中K.lactis 三磷酸甘油醛脱氢酶基因(GAPDH)序列(GenBank 登录号X52871)设计特异性引物GAPDH-F/R,扩增得到941 bp 的片段,连接至质粒pMD 19-T simple,转化E.coli JM109 感受态细胞,挑取阳性转化子,委托上海生工公司测序,得到标准质粒pT-GAPDH。AMPD 基因的标准质粒为pT-AMPD。
1.2.4 GAPDH 基因和AMPD 基因RT-QPCR 反应的标准曲线 以标准质粒pT-GAPDH 和pT-AMPD为标准品,用微量核酸定量仪测定浓度后相应的分子数(拷贝数)为

式中:m 为DNA 质量,g;L 为DNA 长度,bp。
根据标准品每微升质粒溶液所含的拷贝数,对其进行10 倍梯度稀释后用作RT-QPCR 反应的模板,其中pT-GAPDH 标准品分别稀释至103、104、105、106、107个拷贝/μL 的样品,pT-AMPD 标准品分别稀释至102、103、104、105、106个拷贝/μL的样品。根据GAPDH 和AMPD 基因序列,分别设计RTQPCR 反应引物RT-GAPDH-F/R 和RT-AMPD-F/R,扩增产物大小为144,142 bp。反应体系(10 μL):SYBR Premix ExTaqTMII PCR buffer 5 μL,模板0.5 μL,上下游引物各0.3 μL,灭菌的dd H2O 3.9 μL;扩增条件:95 ℃30 s;95 ℃5 s,60 ℃30 s,40 个循环。每个浓度设置3 个平行反应。以起始模板中质粒拷贝数的对数为横坐标,不同浓度梯度下的Ct 值为纵坐标,建立GAPDH 和AMPD 标准曲线。RTQPCR 的扩增效率为

式中:S 为标准曲线的斜率。
1.2.5 AMPD 基因拷贝数的检测 提取重组菌染色体基因组并稀释至40~60 ng/μL 后作为模板,与上述测定标准曲线的RT-QPCR 反应同步进行,得到重组菌基因组中GAPDH 和AMPD 基因的Ct 值,分别代入标准曲线即可得到基因的初始拷贝数,因K.lactis GG799 为单倍体菌株,且GAPDH 在基因组中为单拷贝,AMPD 基因的拷贝数为

式中:CopyA为目的基因起始模板拷贝数;CopyG为GAPDH 基因起始模板拷贝数。
1.2.6 重组菌AMP 脱氨酶酶活测定 AMP 脱氨酶酶活定义:在pH 6.0,60 ℃条件下,反应液于波长265 nm 处吸光值每分钟改变0.001 为酶的一个活力单位。

其中:△A265为试验组与对照组在265 nm 处吸光值的差值;K 为待测酶液的稀释倍数;T 为反应时间,min。
AMP 脱氨酶的酶活参照文献[25]的方法采用紫外分光光度计检测:在试管中加入3 mL 反应底物后置于60 ℃水浴锅中保温5 min,加入100 μL 稀释适当倍数的待测酶液并充分混匀,立即计时,反应15 min,加入3 mL 10%(v/v)高氯酸溶液终止反应。对照试验方法同上,反应底物60 ℃保温后冰水预冷,加3 mL 10%(v/v)的高氯酸后再加100 μL 待测酶液混匀。紫外分光光度计于265 nm 处,用光程10 mm 的石英比色皿测定吸光值(以去离子水校零)。
YEPD 平板活化重组菌,挑取单菌落接种于YEPD 培养基,30 ℃,200 r/min 培养36 h 后转接至50 mL YEPD 培养基,使菌体初始OD600在0.5 左右,30 ℃,200 r/min 摇瓶培养120 h,测定发酵上清液AMP 脱氨酶酶活,设置3 个平行。
1.2.7 重组菌株质粒稳定性测定 YEPD 平板活化重组菌,挑取单菌落接种于20 mL YCB 培养基(100 mL 摇瓶装液量为20 mL),30 ℃,200 r/min 培养16 h,转接至20 mL 新鲜的YEPD 培养基,30 ℃,200 r/min 培养12 h,测定菌液OD600,记为N,转接至新鲜YEPD 培养基中,并控制菌体初始OD600在0.1 左右,每隔12 h 重复此操作,共转接10 次至酵母总繁殖时间为120 h,取适量菌体用无菌水稀释后分别涂布YCB 及YEPD 平板,30 ℃培养3 d,计相应平板上的菌落数,分别记为A1和A2。酵母繁殖世代及质粒稳定性计算公式如下

式中:n 为繁殖世代;N:菌液OD600值;A1为YCB 平板菌落数;A2为YEPD 平板菌落数。
2 结果与分析
2.1 重组表达载体pTRGA-amdS 的构建
以质粒pT-AMPD 为模板PCR 扩增得到AMPD 片段大小为1 476 bp,凝胶电泳显示在1.1~1.7 kb 有单一核酸条带,测序后与目的序列进行BLAST 比对,一致性达到100%,将其连接至载体pKLAC1,酶切验证(图1),构建重组质粒pKLAC1-AMPD。以该质粒为模板,PCR 扩增分别获得4.3 kb的APMD 基因表达框(pLAC4-αMF-AMPD-tLAC4)和2.6 kb 的筛选标记基因表达框(pADH2-amdSTT),连接载体pMD 19T-simple,经测序及BLAST比对表明成功构建质粒pTLA 和pT-amdS。
采用强启动子是实现外源基因高效表达的另一途径,最常用的启动子包括S.cerevisiae 的磷酸甘油酸激酶基因(PGK)启动子和酸性磷酸酶基因(PHO5)启动子,K.lactis 的β-半乳糖苷酶基因(LAC4)启动子[3]。在实验室前期启动子研究工作中,发现S.cerevisiae 来源的pScGAL7 性能优于K.lactis 自身来源的pLAC4(数据他处发表),因此以pScGAL7(301 bp)调控AMPD 的表达,构建质粒pTGA。
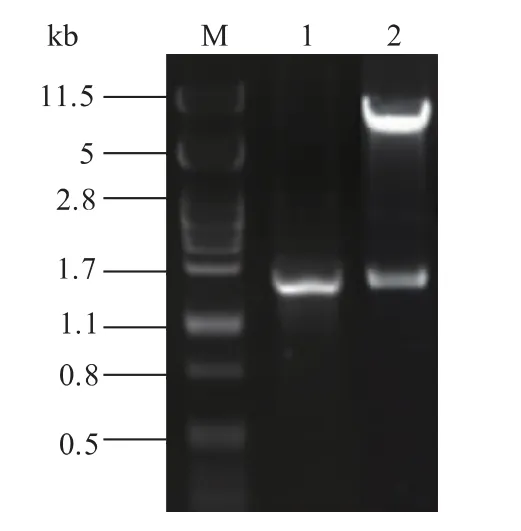
图1 质粒pKLAC1-AMPD 的酶切验证Fig.1 Enzymatic digestion of plasmid pKLAC1-AMPD
根据1.2.1 节所述方法,依次构建质粒pTGAamdS 和pTR,Kpn I 酶切后连接上述两个质粒得重组表达载体pTRGA-amdS,具体构建流程见图2,酶切验证见图3。
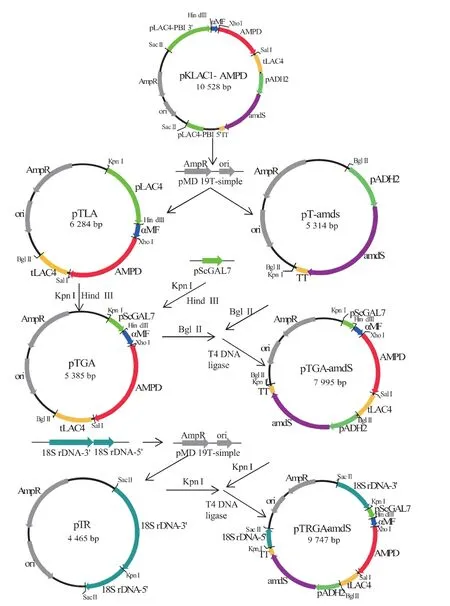
图2 重组表达载体pTRGA-amdS 的构建Fig.2 Construction of recombinant expression vector pTRGA-amdS
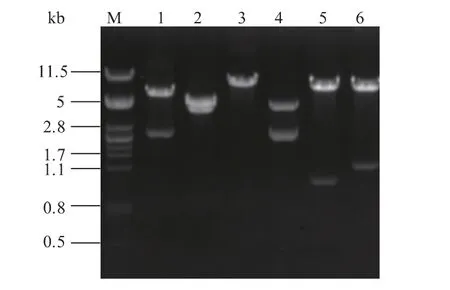
图3 重组表达载体pTRGA-amdS 酶切验证Fig.3 Enzymatic digestion of recombinant expression vector pTRGA-amdS
2.2 转化子的筛选及验证
重组载体pTRGA-amdS 经Sac II 酶切线性化后电转化K.lactis GG799,与基因组中18S rDNA发生同源重组,将AMPD 基因整合到染色体中,α因子信号肽(α-Mating Factor,αMF)介导下实现AMP 脱氨酶的分泌表达,amdS 基因编码的乙酰胺酶能将培养基中的乙酰胺转化为K.lactis 可利用的氮源,只有正确整合的重组菌才能在YCB 培养基中生长,30 ℃培养3~4 d,挑取菌落明显的转化子,提取染色体基因组,设计特异性引物Int-F/R,阳性转化子PCR 扩增片段大小为2 032 bp,而原始菌株无PCR 产物(图4),筛选得到10 株阳性重组菌,-70℃甘油管保藏待用。

图4 重组菌K .lactis/pTRGA-amdS 的PCR 验证Fig.4 Integrated identification of recombinant K .lactis/pTRGA-amdS
2.3 重组菌AMPD 基因拷贝数的测定
以基因组中单拷贝的GAPDH 为内参基因,PCR 扩增得到941 bp 的目的片段,测序结果与K.lactis NRRL Y-1140 的GAPDH 基因序列进行BLAST 比对,一致性达到99.7%,构建标准质粒pTGAPDH,Xho I 和Bgl II 酶切验证(图5)。采用RTQPCR 建立GAPDH 和AMPD 基因的标准曲线见图6。可知,GAPDH 和AMPD 基因的熔解曲线均为单一峰,表明反应过程中未出现非特异性扩增,特异性良好,标准曲线分别为
y=-3.584 7x+39.988 和y=-3.357 3x+37.677,
相关系数(R2)分别为0.998 4 和0.999 1,扩增效率分别为0.9 和0.985 4,符合RT-QPCR 绝对定量扩增效率的要求。以稀释后的阳性重组菌染色体DNA为模板进行RT-QPCR 反应,GAPDH 和AMPD 基因的Ct 值如表3 所示,通过标准曲线计算即可得到反应体系中GAPDH 和AMPD 基因的初始拷贝数,可得10 株重组菌的AMPD 基因拷贝数从1~3 个。
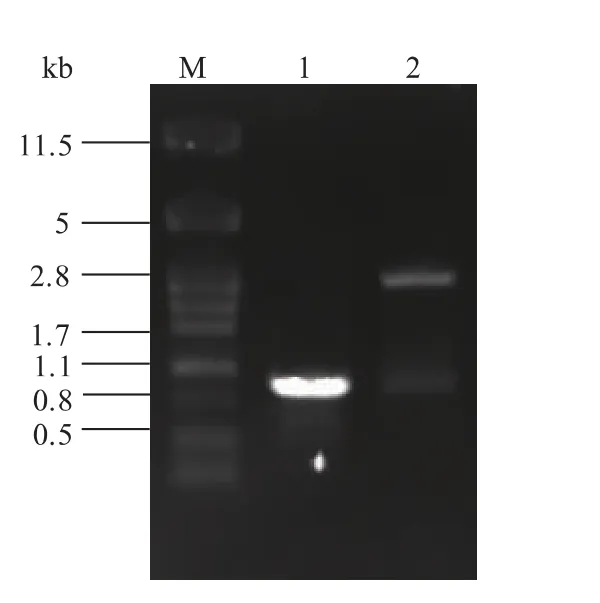
图5 质粒pT-GAPDH 的酶切验证Fig.5 Enzymatic digestion of plasmid pT-GAPDH
K.lactis 染色体中存在68 个rDNA 串联重复序列,每个单元由转录区和非转录区组成,以rDNA序列为同源重组的整合位点兼具高效和稳定的优点而备受青睐。Lin 等[27]利用2.7 kb 的rDNA 序列、URA3 缺陷型筛选标记介导葡糖淀粉酶在S.cerevisiae 中整合表达,拷贝数为25~140。Wery 等[20]以3 kb rDNA 序列同源重组并通过G418 筛选得到50个拷贝的重组红法夫酵母(P.rhodozyma)。Klabunde 等[18]以H.polymorpha 来源的2.4 kb 的25SNTS1-5S-NTS2 为同源重组位点构建了多宿主适用的整合型载体,拷贝数为30~40。但是Steinborn 等[21]以A.adeninivoras 为宿主菌,选取H.polymorpha 来源的rDNA 不同位置的序列构建了一系列重组载体,均得到单拷贝整合重组菌。相似的是,Wartmann等[22]研究以A.adeninivoras rDNA 为整合位点时发现重组菌均以单拷贝或低拷贝整合。研究发现,rDNA 单元中整合位点的位置、载体的大小会影响整合拷贝数及稳定性。目前对于rDNA 整合的分子机制尚不清楚,推测HOT 序列可能与整合稳定性相关[28],该序列是具有重组刺激活性的顺式作用元件,能够促进或激活遗传基因的交流,rDNA 中存在2个HOT 序列,分别位于35S rDNA 前体5’端转录区域的上游和3’ 端的下游,此外,rDNA 中存在的Fob1 因子、Sir2p、RAD52 因子和MRX 复合体因子等重要功能元件均会影响整合的效率和稳定性[6,18]。另外,缺陷型筛选标记或弱启动子的选择下形成的强选择压力易于获得高拷贝重组菌。本研究以完整的18S rDNA 序列为整合位点、amdS 为筛选标记构建重组载体,重组菌整合拷贝数为1~3。在此基础上,可以通过改变整合位点在rDNA 的位置以及利用缺陷型启动子或弱启动子调控amdS 基因来筛选高拷贝重组菌。

图6 GAPDH 和AMPD 基因标准曲线的建立Fig.6 Construction of standard curve of GAPDH and AMPD gene
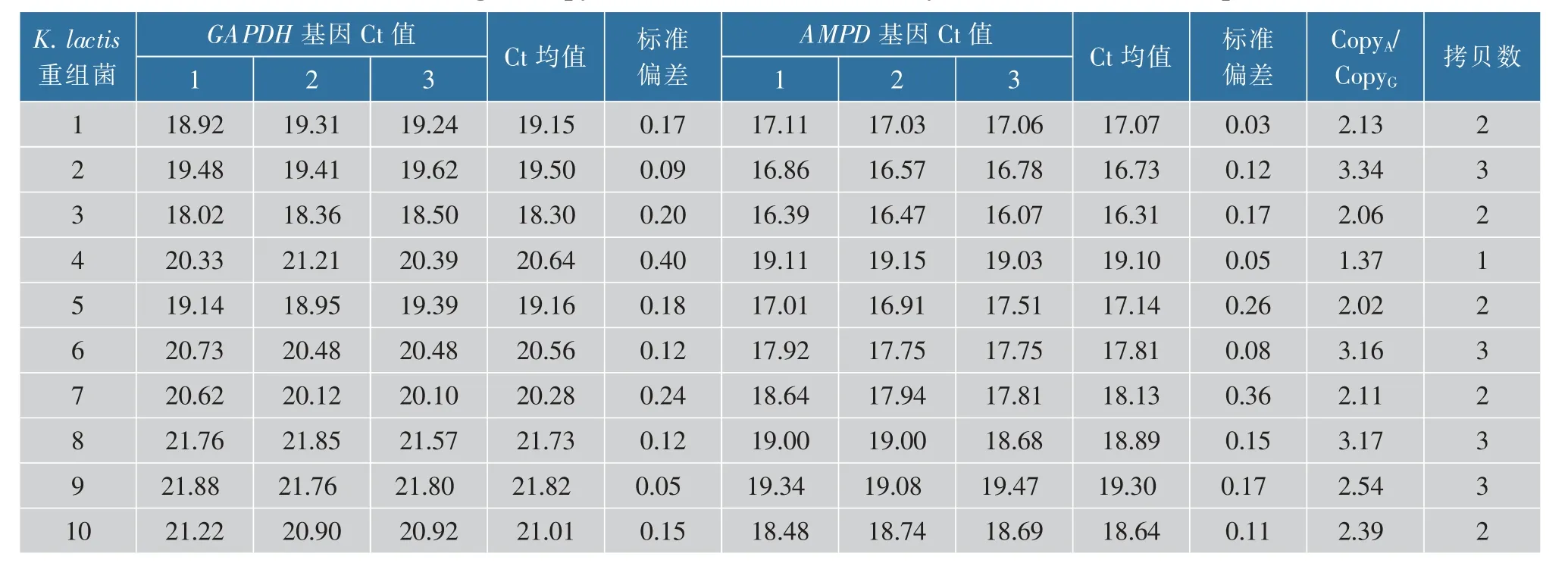
表3 实时荧光定量PCR 检测重组菌AMPD 基因的整合拷贝数Table 3 Estimated AMPD gene copy number of recombinants by real-time fluorescent quantitative PCR
2.4 重组菌AMP 脱氨酶酶活测定
2.4.1 重组菌AMPD 基因拷贝数与AMP 脱氨酶酶活之间的关系 将含不同AMPD 基因整合拷贝的重组菌接种于YEPD 培养基中,测定发酵上清液重组AMP 脱氨酶酶活,结果如图8 所示。重组菌基因组中AMPD 基因拷贝数的差异影响AMP 脱氨酶的表达水平,在同样发酵条件下,只含1 个AMPD 基因拷贝的重组菌发酵上清液AMP 脱氨酶酶活性最低,含3 个AMPD 基因拷贝的重组菌酶活性最高,达到(444.44±23.15)U/mL,比前者提高了88%,含2个AMPD 基因拷贝的重组菌酶活介于1 个和3 个拷贝的重组菌,表明在一定范围内随着拷贝数的增加,重组菌的酶活相应提高,与Marx[9]等研究结果一致,因此,提高目的基因在宿主菌株中的拷贝数是提高外源蛋白表达量的有效途径。虽然本试验中重组菌拷贝数较低,但在强启动子pScGAL7 的调控下外源基因仍得到高效表达。

图7 重组菌AMP 脱氨酶酶活性与AMPD 基因拷贝数的关系Fig.7 Relationship between AMP deaminase activity and the copy number of the AMPD gene in recombinants
2.4.2 培养基成分对重组菌生长和产酶的影响重组菌K.lactis/pTRGA-amdS 中AMPD 基因的表达受pScGAL7 调控,该启动子为非严格诱导型启动子,由乳糖或半乳糖诱导,但不完全被葡萄糖抑制。在诱导和非诱导培养基中分别测定重组菌菌体生长量和发酵上清液AMP 脱氨酶酶活,结果如图9所示。在K.lactis 中,pScGAL7 在缺少诱导物的条件下存在一定量的本底表达,AMP 脱氨酶酶活为(445±11.67)U/mL,诱导条件下,YEPDG 培养基中酶活为(560±20)U/mL,YEPG 培养基中酶活最高,为(590±13.33)U/mL,与非诱导条件下相比分别提高了25.8%和32.6%,而在不同培养基中重组菌菌体生长量相当,进一步表明酶活的提高是由于诱导物存在下启动子调控外源蛋白表达的能力增强,且随着诱导物浓度的增加,酶活相应提高。
2.5 乳酸克鲁维酵母整合表达体系稳定性
在无选择压力条件下,与游离型表达载体相比,整合型表达载体具有在宿主菌基因组中稳定遗传的优点。根据1.2.7 节所述方法,对构建的重组表达载体在细胞内的稳定性进行了测定,重组菌传代58 次后的稳定性为98.59%,表明整合于K.lactis基因组中的外源基因具有良好的遗传稳定性。
3 讨论
以18S rDNA 为同源重组的整合位点构建了适用于K.lactis 的整合型表达载体pTRGA-amdS,并实现了AMP 脱氨酶在宿主菌中的分泌表达。所构建的载体在以下方面具有一定的优势:1)以rDNA为同源重组位点,外源基因整合于宿主细胞基因组中,且在无选择压力条件下稳定遗传;2)线性化重组载体后,同源重组片段去除了E.coli 来源的ori序列和氨苄青霉素抗性基因(Ampr),减小了载体的大小,同时提高了稳定性和生物安全性[12];3)以amdS 为筛选标记,可直接从野生型菌株出发,不需要利用URA、LEU 或TRP 等营养缺陷型筛选标记,省去了营养缺陷型菌株的筛选,并避免了回复突变的问题[2],此外,也降低了使用G418、潮霉素等抗生素在筛选中的成本,不会对环境和人类产生危害;4)在整合拷贝数较低的情况下,强启动子pScGAL7调控外源基因的表达达到较高的胞外蛋白产量,另外,因rDNA 在K.lactis 中基因组中存在70 个左右拷贝,该表达载体也为同时共表达多个外源基因(如酵母分泌途径中的辅助因子或分子伴侣等)提供了可能[7-8]。
与此同时,该表达载体也存在一定的不足之处,即重组菌整合拷贝数较低,推测可能原因一方面是本研究所选取的同源臂序列为完整的18S rDNA,该位点具有高度保守性,且同源重组片段缺少了HOT、Fob1 等重要功能元件,因此影响了整合的效率[6,18],另一方面是调控筛选标记的pADH2 为强启动子,重组菌只需达到较低拷贝即能在YCB 平板上生长,不利于拷贝数的复制。为了避免上述情况,后续试验可以从两个角度出发,一是对K.lactis的rDNA 序列进行分析,选择合适的同源臂序列,二是将amdS 基因前的启动子替换为缺陷型启动子或弱启动子,通过增强选择压力来筛选高拷贝重组菌。
4 结语
利用本研究构建的表达载体能够实现外源基因的稳定表达,为K.lactis 进行分子改造提供了理想工具。因目前对于rDNA 的重要元件、重组的分子机制研究尚不完善,通过不断深入研究,以rDNA 重复单元作为同源重组的整合位点将在基因工程应用中发挥越来越重要的作用。
猜你喜欢
右江民族医学院学报(2022年2期)2022-05-19
河北医学(2021年10期)2021-10-27
中国临床医学影像杂志(2019年6期)2019-08-27
中国生殖健康(2018年1期)2018-11-06
天津农业科学(2018年5期)2018-06-23
中国医药指南(2018年3期)2018-03-23
今日中国·中文版(2017年8期)2017-09-03
中华实验和临床病毒学杂志(2016年3期)2016-08-09
中国医药指南(2015年29期)2015-10-27
植物营养与肥料学报(2011年6期)2011-10-24
