
扶手椅型单壁氮化硼纳米管的尺寸对缬氨酸旋光异构的限域影响∗
2019-01-25 08:20姜春旭杨晓翠绳绍旭白杰丛建民王佐成
中山大学学报(自然科学版)(中英文) 2019年1期
姜春旭,杨晓翠,绳绍旭,白杰,丛建民,王佐成
(1. 白城师范学院理论计算中心,吉林 白城137000; 2. 白城师范学院传媒学院,吉林 白城 137000;3. 白城师范学院物理学院,吉林 白城 137000;4. 白城师范学院生命科学学院,吉林 白城 137000)
缬氨酸(valine, Val)是生命体的必需氨基酸,根据构象的不同,分为S型缬氨酸(S-Val)和R型缬氨酸(R-Val);根据旋光性的不同,分为左旋缬氨酸(L-Val)和右旋缬氨酸(D-Val)。L-Val对生命体具有重要作用,可以促进机体的发育,还可以使神经系统协调[1]。D-Val可控制纤维细胞的生长[2],可用于氯氟戊菊酯的合成[3],也是合成手性抗肿瘤药物的重要中间体[4],其价格比较昂贵。
由于光学纯Val的重要性,学者们在其旋光异构领域做了大量的工作。Cholewinski A J等[2]的研究表明,在羧酸中醛的催化可使Val分子旋光异构。文献[5]和[6]在理论和实验两个方面的研究表明,Val分子在高浓度的质子环境下可以消旋。文献[7-8]的研究表明,水汽环境下水分子簇对Val旋光异构具有催化作用,文献[9]的研究发现,Val在激发态可以实现旋光异构,水溶剂对反应有阻碍作用。文献[10-12]的研究发现,氢氧根离子水分子团簇可催化缬氨酸迅速旋光异构,羟自由基抽取α-H可致缬氨酸迅速损伤,结果表明,生命体内氢氧根离子过量以及强自由基的存在会严重影响健康,同时也说明了光学纯的缬氨酸很难储存。
生命体内纳米环境下缬氨酸的旋光异构,以及对于价格昂贵的光学纯缬氨酸存储的研究,目前鲜见报道。已有研究表明[13-15],限域在纳米管内化学反应的表现不同于裸反应,并且纳米管尺寸的不同对限域其中的化学反应表现出显著不同的影响,寻找一种纳米管,利用其负催化作用储存光学纯缬氨酸具有重要意义。基于此,本工作对扶手椅型单壁氮化硼纳米管(SWBNNT)内缬氨酸分子的旋光异构进行了研究,并期望在理论上寻找到存储光学纯缬氨酸的一维纳米碳材料。
1 模型的选取与计算研究方法
缬氨酸分子的横向线度约为0.580 1 nm,纵向线度约为0.380 7 nm×0.395 1 nm,扶椅型单臂氮化硼纳米管(5,5)[SWBNNT(5,5)]的直径是0.673 2 nm,扶椅型单臂碳纳米管(7,7)[SWCBNNT(7,7)]的直径是0.942 6 nm。Val分子限域在直径小于SWBNNT(5,5)的氮化硼纳米管时,与SWBNNT是化学吸附,而Val分子在直径大于SWBNNT(7,7)的SWBNNT时,SWBNNT的限域影响已不存在,因此本工作把SWBNNT(5,5)、(6,6)和(7,7)作为纳米限域环境。为充分考虑SWBNNT的限域效应,管长取为2.500 nm(截断处的氮原子用氢原子饱和)。
采用量子化学组合的ONIOM方法[16]研究标题反应。将SWBNNT与驻点分子分两层:驻点分子为高层,用量子力学描述,SWBNNT为低层,用分子力学的UFF力场[17]描述;为较好地处理驻点分子与SWBNNT间的长程作用,采用长程校正泛函的CAM-B3LYP[18]方法;基组选用6-31+G(d, p);在ONIOM(CAM-B3LYP/6-31+G(d, p): UFF)水平全参数优化稳定点和过渡态[19]。高层采用微扰论的MP2[20]方法,在ONIOM(MP2/6-311++G(2df, pd): UFF)水平下计算驻点包结物分子的单点能(ESP),吉布斯自由能热校正(Gtc)和单点能相加作为总吉布斯自由能(Gtotal),从而获得反应过程精确的势能剖面。通过内禀反应坐标(IRC)计算[21]确认过渡态。用S-Val表示S型Val分子,S-Val限域在SWBNNT(5,5)的包结物分子用S-Val@SWBNNT(5,5)表示,其他包结物分子用类似方法表示。本工作的计算由Gaussian09软件包[22]完成。
2 研究结果及讨论
最稳定的S-Val分子及其手性对映体构象[10]见图1,可以看出,S-Val的旋光异构,就是α-碳上的质子从纸面外侧迁移到纸面里侧。
文献[8-10]的研究表明,缬氨酸的旋光异构可以在若干个通道实现,α-氢以氨基氮为桥迁移的通道具有优势,本工作仅对优势通道进行讨论。
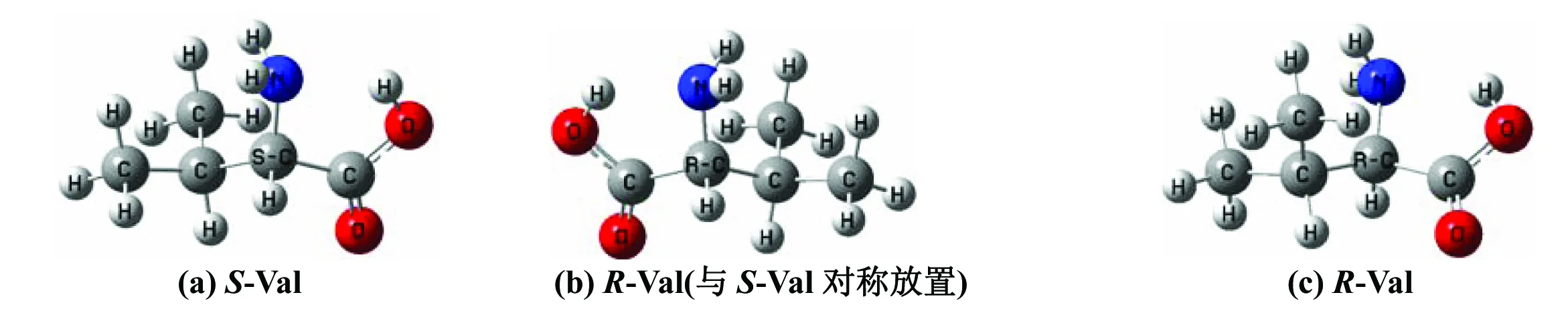
图1 S型与R型缬氨酸分子的几何结构Fig.1 Geometries of S-type and R-type of valine molecules
2.1 S-Val分子限域在SWBNNT(5,5)和SWBNNT(6,6)内的旋光异构
S-Val限域在SWBNNT(5,5)内,α-氢以氨基氮为桥迁移的通道是羧基异构后α-氢再以氨基氮为桥从α-碳向羰基氧迁移,反应历程,见图2,反应过程的吉布斯自由能曲线,见图5。
先是反应物包结物S-Val@SWBNNT(5,5),经190H绕187C-188O旋转的过渡态S-TS1@SWBNNT(5,5)实现构象异构,得到的第一中间体包结物记为S-INT1@SWBNNT(5,5)。此过程,二面角190H—188O—187C—189O从165.83°变为-1.13°,实现羧基从反式平面结构向顺式的构象异构,同时氨基团俯视逆时针旋转,二面角185H—184N—181C—182H从-81.33°变为-107.40°。这时,184N朝向读者的一面也裸露出来,负电荷密度显著增加,容易获得质子;185H和190H向两侧的旋转降低了α-氢向氨基氮迁移的空间位阻。从S-Val@SWBNNT(5,5)到S-TS1@SWBNNT(5,5),二面角190H—188O—187C—189O从165.83°变为-113.56°,二面角185H—184N—181C—182H从-81.33°变为-105.55°,187C—188O键长从0.133 0 nm微增到0.135 4 nm。此过程无断键,只是羟基和氨基旋转,过渡态的能垒不高,只有19.67 kJ·mol-1。与裸反应[10]的50.36 kJ·mol-1相比较明显降低,原因是SWBNNT(5,5)的限域使从反应物到过渡态二面角190H—188O—187C—189O的改变以及187C—188O键长的伸长小于裸反应的情形[10],SWBNNT(5,5)对此基元有明显的限域催化作用。第一中间体与反应物的相对能量是-29.16 kJ·mol-1,相对于S-Val@SWBNNT(5,5)体系显著降低,主要原因是S-INT1的偶极矩(2.436 5)明显小于S-Val的偶极矩(5.552 8),SWBNNT可看作是“无极性溶剂”,无极性溶剂与极性相对小的分子相互作用会使体系能量降低得更多。因此,S-INT1@SWBNNT(5,5)体系能量相对降低的明显,构象稳定。
然后,包结物分子S-INT1@SWBNNT(5,5),经α-氢182H从α-碳181C向氨基氮234N迁移,接着又向羰基氧189O迁移的质子连续迁移及羧基旋转的过渡态TS2@SWBNNT(5,5),构型异构成中间体INT2@SWBNNT(5,5)。从S-INT1@SWBNNT(5,5)到TS2@SWBNNT(5,5)过程,IRC计算表明:181C—182H键长从0.103 2 nm拉伸到0.139 7 nm键断裂,伸长量是0.036 5 nm;181C—184N键长从0.141 5 nm增加到0.157 3 nm,伸长0.015 8 nm;活性中心骨架二面角184N—181C—183C—187C从111.79°变为95.52°,改变16.3°。此过程2个化学键拉伸以及骨架形变均大于裸环境(分别为:0.027 7 nm、0.013 2 nm、11.5°),因此,TS2@SWBNNT(5,5)产生了较高的能垒,能垒值是318.41 kJ·mol-1(远高于裸环境此基元反应的能垒268.93 kJ·mol-1)。这与以往研究[13-15]得到的纳米管限域正催化丙氨酸旋光异构过程的氢迁移反应截然不同,原因是缬氨酸的R-基是2个甲基和一个次甲基,R-基占据较大的空间,导致其从S-INT1@SWBNNT(5,5)到TS2@SWBNNT(5,5)过程化学键拉伸以及骨架形变显著增大,这与分子筛同一空穴对不同构象的分子的择形催化作用相似。
接下来,INT2@SWBNNT(5,5)经和TS2@SWBNNT(5,5)准对称的过渡态TS3_1@SWBNNT(5,5),实现了182H从羰基氧189O向氨基氮184N和186H在纸面里从氨基氮184N向α-碳181C的协同迁移,构型异构成R型产物包结物P1_R-Val@SWBNNT(5,5),S-Val实现旋光异构。从INT2@SWBNNT(5,5)到TS3@SWBNNT(5,5)过程,189O—182H键长从0.092 9 nm拉伸到0.159 2 nm键断,184N—186H键长从0.099 8 nm增加到0.122 8 nm,2个化学键的大幅拉伸断裂需要较高的能量,因此,TS3@SWBNNT(5,5)产生的能量也很高,是225.29 kJ·mol-1。P1_R-Val@SWBNNT(5,5)又可以经过羧基内质子迁移的过渡态R-TS4@SWBNNT(5,5),构象异构成P2_R-Val@SWBNNT(5,5)。从P1_R-Val@SWBNNT(5,5)到R-TS4@SWBNNT(5,5)过程,188O—190H键长从0.097 2 nm拉伸到0.127 0 nm键断,拉伸幅度不是很大,因此,R-TS4@SWBNNT(5,5)产生的能垒不是很高,只有98.52 kJ·mol-1。

图2 S-Val限域在SWBNNT(5,5)内的旋光异构历程及驻点构象Fig.2 Optical isomers process and stationary point conformation of S-Val confined in SWBNNT(5,5)
过渡态TS2@SWBNNT(5,5)连接的产物中间体是INT2@SWBNNT(5,5),令人费解。从TS2@SWBNNT(5,5)到INT2@SWBNNT(5,5)的IRC计算说明:182H从过渡态开始,先迁移到184N上,然后经无势垒过程向189O迁移并伴随着羧基右视顺时针旋转。在路径上选取的几个代表性结构,说明了TS2@SWBNNT(5,5)到INT2@SWBNNT(5,5)的简要历程,见图3。IRC计算表明:从中间体反应物包结物INT2@SWBNNT(5,5)到过渡态包结物TS3@SWBNNT(5,5)的历程与TS2@SWBNNT(5,5)到INT2@SWBNNT(5,5)的历程基本相反,不再赘述。
S-Val限域在SWBNNT(6,6)内,α-氢以氨基氮为桥迁移有两个通道a和b,反应历程见图4,旋光异构反应的吉布斯势能曲线,见图5。
对于a通道:先是限域在SWBNNT(6,6)内缬氨酸的包结物分子S-Val@SWBNNT(6,6),经羟基旋转的过渡态a_S-TS1@SWBNNT(6,6),构象异构成为中间体a_S-INT1@SWBNNT(6,6)。190H—188O—187C—189O二面角从161.44°变为-2.24°,实现了羧基从反式向顺式平面结构异构,减小了α-氢182H向氨基氮184N迁移的空间位阻。从S-Val@SWBNNT(6,6)到a_S-TS1@SWBNNT(6,6),二面角190H—188O—187C—189O从161.44°变为-108.02°,二面角185H—184N—181C—182H从-84.10°变为-173.06°,2个二面角的旋转所需能垒不高,过渡态a_S-TS1@SWBNNT(6,6)产生的能垒是24.29 kJ·mol-1。

图3 TS2@SWBNNT(5,5)到INT2@SWBNNT(5,5)的简明反应过程Fig.3 The concise reaction process of TS2@SWBNNT(5,5) to INT2@SWBNNT(5,5)
产物包结物相对于反应物包结物的能量是-39.65 kJ·mol-1,说明a_S-INT1@SWBNNT(6,6)的能量相对于S-Val@SWBNNT(6,6)显著降低,其原因同于a_S-INT1@SWBNNT(5,5)的能量相对于S-Val@SWBNNT(5,5)显著降低,不再赘述。
然后,a_S-INT1@SWBNNT(6,6),经过渡态a_TS2@SWBNNT(6,6)构型异构,形成中间体包结物a_INT2@SWBNNT(6,6)。从a_S-INT1@SWBNNT(6,6)到a_TS2@SWBNNT(6,6)过程,181C—182H键长从0.107 7 nm拉伸到0.135 6 nm键断裂,伸长量是0.027 9 nm;181C—184N键长从0.144 8 nm增加到0.154 4 nm,伸长0.009 6 nm;反应活性中心骨架二面角184N—181C—183C—187C从113.68°变为125.55°,改变11.87°,这几个参数的变化基本同于裸反应;标志羧基旋转的二面角181C—187C—189O—188O从77.23°变为26.27°,改变较大,远远大与裸反应[10]。此过程化学键的拉伸和骨架变化总体大与裸反应,但小于S-INT1@SWBNNT(5,5)到TS2@SWBNNT(5,5)的情况,因此,a_TS2_1@SWBNNT(6,6)产生了高于裸环境但低于a_TS2@SWBNNT(5,5)的能垒,能垒是306.93 kJ·mol-1。
接着是a_INT2@SWBNNT(6,6)经过渡态a_TS3@SWBNNT(6,6),实现了185H在纸面里从氨基氮184N向α-碳181C的迁移,构型异构成为第一产物包结物a_P1_R-Val@SWBNNT(6,6),S-Val_1实现了旋光异构。从a_INT2@SWBNNT(6,6)到a_TS3@SWBNNT(6,6)过程,键长184N—185H从0.102 2 nm拉伸到0.113 7 nm,键断,181C—184N键长从0.145 6 nm增加到0.153 8 nm,两个化学键的拉伸幅度远小于从a_S-INT1@SWBNNT(6,6)到a_TS2@SWBNNT(6,6)过程,需要的能量也会少许多。又活性中心骨架二面角184N—181C—183C—187C从125.65°变为-128.92°,α-碳181C从空配向满配(sp2杂化向sp3杂化)过渡,释放能量。因此,a_TS3@SWBNNT(6,6)产生的能垒不高,只有74.22 kJ·mol-1。
最后,a_P1_R-Val@SWBNNT(6,6)经过渡态a_R-TS4@SWBNNT(6,6),实现了羧基从顺式向反式平面结构的构象异构,形成第二产物a_P2_R-Val@SWBNNT(6,6)。此基元反应只是二面角异构,能垒较低,只有47.41 kJ·mol-1。
对于b通道:首先,反应物包结物S-Val@SWBNNT(6,6),经过渡态b_TS1@SWBNNT(6,6),构型异构成第1中间体包结物b_INT1@SWBNNT(6,6)。在此基元,实现了α-氢182H从α-碳181C向氨基氮184N迁移与羟基旋转的协同过程,完成了a通道1和2两个基元的“工作”,b_TS1@SWBNNT(6,6)产生的内禀能垒是306.42 kJ·mol-1。计算表明,b_INT1@SWBNNT(6,6)全同于a_INT2@SWBNNT(6,6),接下来的反应机理不在赘述。b_TS1@SWBNNT(6,6)的偶极矩(5.437 1)大于a_S-TS1@SWBNNT(6,6)的偶极矩(4.159 8),电荷分离程度高,稳定性前者小于后者,b_TS1@SWBNNT(6,6)处在势能面较高的位置上。但它们产生的内禀能垒却差不多,原因是S-Val@SWBNNT(6,6)与a_S-INT1@SWBNNT(6,6)相比处在势能面较高的位置上。

图4 S-Val限域在SWBNNT(6,6)内的旋光异构历程及驻点构象Fig.4 Optical isomers process and stationary point conformation of S-Val confined in SWBNNT(6,6)
从图5可以看出:S-Val限域在SWBNNT(5,5)旋光异构决速步骤是第2基元,决速步能垒是318.41 kJ·mol-1,产物会以P1_R-Val@SWBNNT(5,5)和P2_R-Val@SWBNNT(5,5)两种构象共存,后者的分布要高些。S-Val限域在SWBNNT(6,6)在a通道旋光异构的决速步骤也是第2基元,决速步能垒是306.93 kJ·mol-1,在b通道旋光异构的决速步骤是第1基元,决速步能垒是306.42 kJ·mol-1,这两个通道的优劣不分伯仲。这两个通道的产物都会以P1_R-Val@SWBNNT(6,6)和P2_R-Val@SWBNNT(6,6)两种构象共存,前者的分布要远高于后者。
S-Val限域在SWBNNT(5,5)和SWBNNT(6,6)旋光异构的决速步能垒均高于裸环境的268.93 kJ·mol-1[10]许多,说明SWBNNT(5,5)和SWBNNT(6,6)会很好地阻碍S-Val的旋光异构,均可以作为光学纯缬氨酸的理想“存储器”,比较而言,SWBNNT(5,5)更理想。
2.2 S-Val分子限域在SWBNNT(7,7)内的旋光异构
研究表明,限域在SWBNNT(7,7)内的S-Val,α-氢以氨基氮为桥迁移的旋光异构也存在相似于在SWBNNT(6,6)内的两个通道a和b,a通道在竞争中具有优势。为节省篇幅,仅对优势通道a的旋光异构进行讨论。图6为旋光异构历程,图7为吉布斯自由能剖面。
首先是反应物包结物分子S-Val@SWBNNT(7,7),经过渡态S-TS1@SWBNNT(7,7),构象异构,形成中间体S-INT1@SWBNNT(7,7)。S-INT1@SWBNNT(7,7)的羧基为顺式结构,并且二面角185H—184N—181C—182H变为-69.77°,使α-氢182H向氨基氮184N迁移的位阻显著降低。同时氨基氮184N朝向读者的一面基本裸露出来,负电荷密度增加,也是其获得质子的能力增加。从S-Val@SWBNNT(7,7)到S-TS1@SWBNNT(7,7)过程,二面角190H—188O—187C—189O从177.79°变为-91.06°,二面角185H—184N—181C—182H从-45.31°变为-57.35°,187C—188O键长从0.133 7 nm拉伸到0.137 8 nm,构象变化基本同于裸反应[10],SWBNNT(7,7)的限域作用基本消失。S-TS1@SWBNNT(7,7)产生的能垒是39.72 kJ·mol-1,基本同于裸反应[10]。
然后,中间体反应物S-INT1@SWBNNT(7,7),经过渡态TS2@SWBNNT(7,7)构型异构,形成第二中间体产物包结物INT2@SWBNNT(7,7)。从S-INT1@SWBNNT(7,7)到TS2@SWBNNT(7,7)过程,181C—182H键长从0.109 4 nm
拉伸到0.136 4 nm键断裂,伸长量是0.027 0 nm;181C—184N键长从0.145 1 nm增加到0.156 8 nm,伸长0.011 7 nm;活性中心骨架二面角234N—231C—233C—237C从125.86°变为137.96°。构象几何参数变化基本同于裸反应[10],说明SWBNNT(7,7)的限域作用消失,过渡态TS2@SWBNNT(7,7)产生的能垒是262.81 kJ·mol-1,在误差允许的范围内,可认为同于裸反应的268.93 kJ·mol-1[10]。由于INT2@SWBNNT(7,7)的正负电荷中心分离,并且α-碳181C是sp2杂化,处于非满配状态,导致INT2@SWBNNT(7,7)体系的较高(相对于S-Val@SWBNNT(7,7)),处在势能面较高的位置。
接着是INT2@SWBNNT(7,7)作为中间体反应物,经过渡态TS3@SWBNNT(7,7),质子化氨基上的185H在纸面里从184N向181C(α-碳)的迁移,构型异构为R型中间体产物R-INT3@SWBNNT(7,7),此时,S-Val已经实现旋光异构。从INT2@SWBNNT(7,7)到TS3@SWBNNT(7,7)过程,184N—185H键长从0.102 7 nm拉伸到0.116 1 nm键断,181C—184N键长从0.147 8 nm增加到0.156 8 nm。两个化学键的断裂需要一定的能量,但活性中心骨架二面角184N—181C—183C—187C从9.37°变为137.49°,α-碳181C从sp2杂化向sp3杂化过渡,释放能量,因此,TS3@SWBNNT(7,7)产生的能量不是很高,只有105.12 kJ·mol-1。
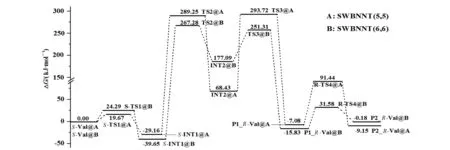
图5 S-Val限域在A和B内旋光异构反应的吉布斯自由能剖面Fig.5 Gibbs free energy profile of optical isomers reaction of S-Val confined in A and B

图6 S-Val限域在SWBNNT(7,7)内的旋光异构历程及驻点结构Fig.6 Optical isomers process and stationary point conformation of S-Val confined in SWBNNT(7,7)
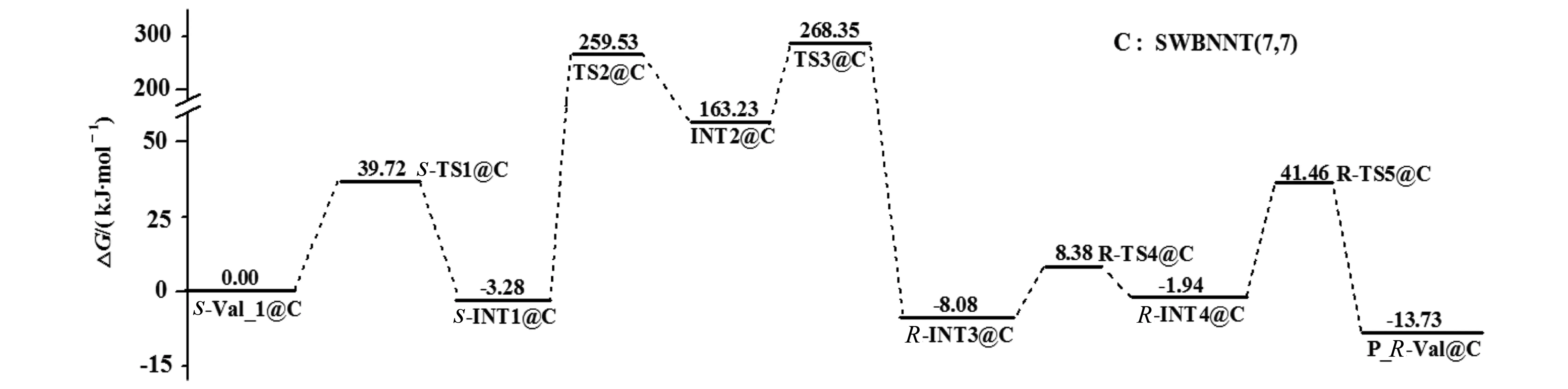
图7 S-Val限域在SWBNNT(7,7)内旋光异构反应的吉布斯自由能剖面Fig.7 Gibbs free energy profile of optical isomers reaction of S-Val confined in SWBNNT(7,7)
再接下来是,R-INT3@SWBNNT(7,7)经氨基左右翻转的过渡态a_R-TS4@SWBNNT(7,7),构象异构成产物包结物R-INT4@SWBNNT(7,7)。此基元无断键,能垒只有16.42 kJ·mol-1。
最后,R-INT4@SWBNNT(7,7)经过渡态a_R-TS4_1@SWBNNT(7,7),实现羧基异构,构象异构成产物包结物P_R-Val@SWBNNT(7,7)。此基元相似于第一基元,反应过程无断键,能垒是43.40 kJ·mol-1。P_R-Val具有氨基和羧基间的分子内单氢键,因此包结物P_R-Val@SWBNNT(7,7)体系构象稳定。
从图7可以看出:S-Val旋光异构的决速步是第2基元,内禀能垒是262.81 kJ·mol-1,与裸反应的决速步能垒268.93 kJ·mol-1[10]相差无几。说明SWBNNT(7,7)的限域对S-Val的旋光异构已经没有影响。
3 结 论
氨基氮作为质子转移桥梁是缬氨酸分子旋光异构反应的优势通道。缬氨酸限域在SWBNNT(5,5)内时,只有一个路径,决速步的内禀能垒是318.41 kJ·mol-1;缬氨酸限域在SWBNNT(6,6)内时,有两条路径,决速步的内禀能垒分别是306.42和306.93 kJ·mol-1。缬氨酸限域在SWBNNT(7,7)时,旋光异构决速步的内禀能垒是262.81 kJ·mol-1,与裸反应相差无几。结果表明:SWBNNT(5,5)和SWBNNT(6,6)的限域对缬氨酸分子旋光异构具有明显的负催化作用,可以作为光学纯缬氨酸理想的存储器;缬氨酸限域在SWBNNT(7,7)以及孔径更大的纳米管时,纳米管的限域效应消失。
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
北京航空航天大学学报(2022年5期)2022-06-06
物理学报(2022年10期)2022-06-04
原子与分子物理学报(2022年3期)2022-03-05
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
教育教学论坛(2017年14期)2017-04-20
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
中学化学(2015年8期)2015-12-29
